

Glioblastoma Stem Cells at the Nexus of Tumor Heterogeneity, Immune Evasion, and Therapeutic Resistance

Abstract
:1. Introduction
2. Limitations of Current Therapeutic Strategies
3. The Concept of Cancer Stem Cells in Glioblastoma
4. Characteristics of GBM Stem Cells (GSCs)
4.1. Identification and Isolation of GSCs
4.2. Biological Properties of GSCs
4.3. Signaling Pathways Focusing on GSCs
5. GSCs in Tumor Initiation and Progression
5.1. Role in Tumor Heterogeneity
5.2. Angiogenesis and the Vascular Niche
5.3. Immune Modulation
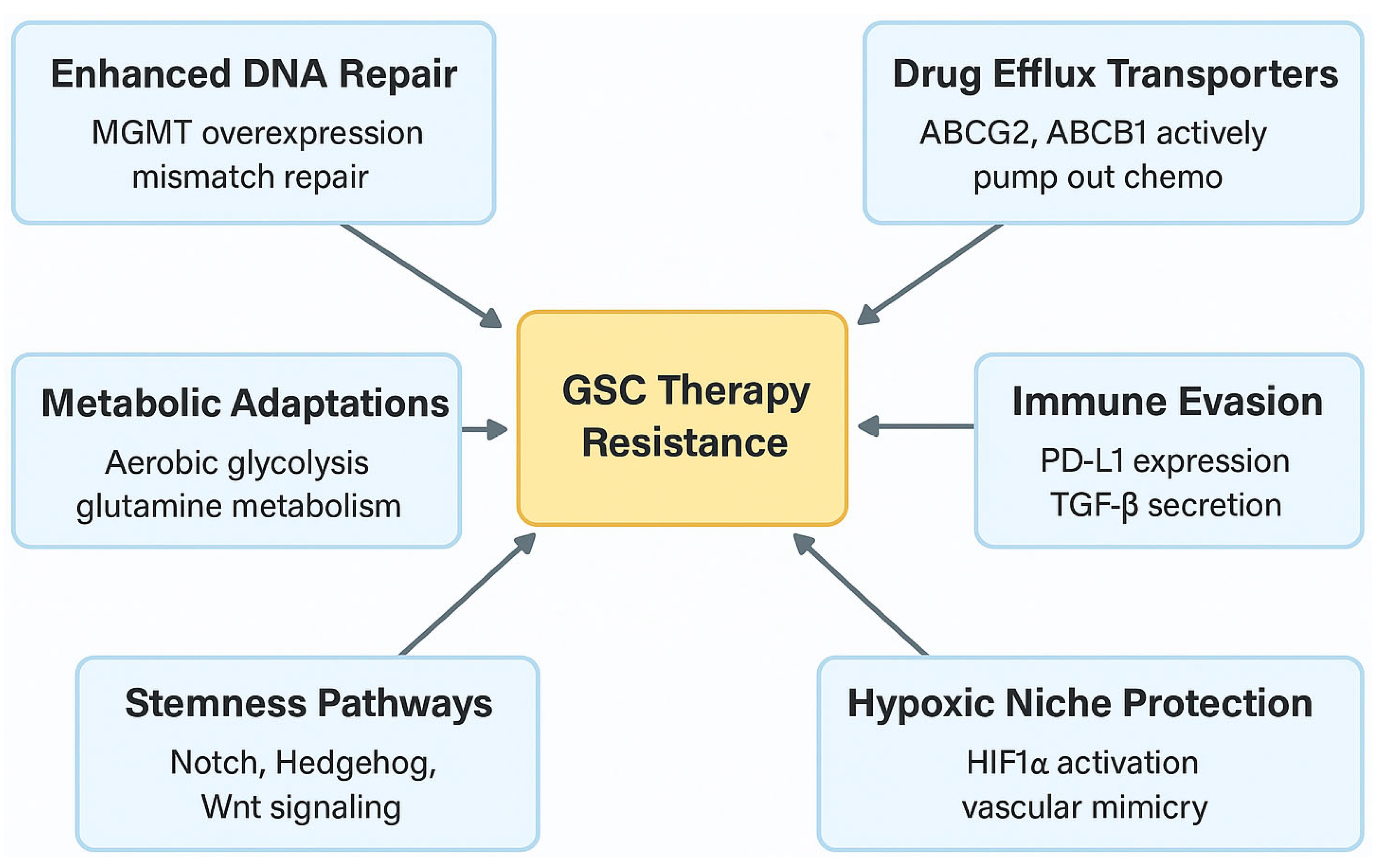
6. GSCs and Therapy Resistance
6.1. Mechanisms of Chemotherapy Resistance
6.2. Metabolic Adaptations
7. Therapeutic Strategies Targeting GSCs
7.1. Targeting Surface Markers
7.2. Immunotherapeutic Approaches
8. Challenges in Targeting GSCs
8.1. Marker Heterogeneity and Specificity
8.2. Tumor Microenvironment Influence
8.3. Therapeutic Resistance and Adaptation
9. Future Perspectives
9.1. Advancements in GSC Research Models
9.2. Single-Cell Omics and Personalized Medicine
9.3. Combination Therapies
10. Discussion and Future Directions
11. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Karbhari, N.; Campian, J.L. Therapeutic Targets in Glioblastoma: Molecular Pathways, Emerging Strategies, and Future Directions. Cells 2025, 14, 494. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes. Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes. Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef]
- Sanai, N.; Berger, M.S. Surgical oncology for gliomas: The state of the art. Nat. Rev. Clin. Oncol. 2018, 15, 112–125. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Senft, C.; Bink, A.; Franz, K.; Vatter, H.; Gasser, T.; Seifert, V. Intraoperative MRI guidance and extent of resection in glioma surgery: A randomised, controlled trial. Lancet Oncol. 2011, 12, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Sener, U.T.; Sulman, E.P.; Sarkaria, J.N. Temozolomide use in elderly patients with MGMT promoter unmethylated glioblastoma: Is it finally time to dismount a dead horse? Neuro Oncol. 2024, 26, 1876–1877. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, D.; Savage, J.; Patel, A.; Collinson, F.; Mant, R.; Boele, F.; Brazil, L.; Meade, S.; Buckle, P.; Lax, S.; et al. A randomised phase II trial of temozolomide with or without cannabinoids in patients with recurrent glioblastoma (ARISTOCRAT): Protocol for a multi-centre, double-blind, placebo-controlled trial. BMC Cancer 2024, 24, 83. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.C.; Bjerkvig, R. Molecular mechanisms of temozolomide resistance in glioblastoma multiforme. Expert. Rev. Anticancer. Ther. 2012, 12, 635–642. [Google Scholar] [CrossRef]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Rich, J.N.; Bao, S. Chemotherapy and cancer stem cells. Cell Stem Cell 2007, 1, 353–355. [Google Scholar] [CrossRef]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer stem cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef]
- Vescovi, A.L.; Galli, R.; Reynolds, B.A. Brain tumour stem cells. Nat. Rev. Cancer 2006, 6, 425–436. [Google Scholar] [CrossRef]
- Lathia, J.D.; Liu, H. Overview of Cancer Stem Cells and Stemness for Community Oncologists. Target. Oncol. 2017, 12, 387–399. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Clarke, M.F. Therapeutic implications of the cancer stem cell hypothesis. Semin. Radiat. Oncol. 2009, 19, 78–86. [Google Scholar] [CrossRef]
- Safa, A.R. Drug and apoptosis resistance in cancer stem cells: A puzzle with many pieces. Cancer Drug Resist. 2022, 5, 850–872. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Moore, N.; Lyle, S. Quiescent, slow-cycling stem cell populations in cancer: A review of the evidence and discussion of significance. J. Oncol. 2011, 2011, 396076. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Emir, S.M.; Karaoğlan, B.S.; Kaşmer, R.; Şirin, H.B. Hunting glioblastoma recurrence: Glioma stem cells as retrospective targets. Am. J. Physiol. Cell Physiol. 2025, 328, C1045–C1061. [Google Scholar] [CrossRef]
- Eyler, C.E.; Rich, J.N. Survival of the fittest: Cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 2008, 26, 2839–2845. [Google Scholar] [CrossRef]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Hide, T.; Dirks, P.B. Cancer stem cells in nervous system tumors. Oncogene 2004, 23, 7267–7273. [Google Scholar] [CrossRef]
- Wang, J.; Sakariassen, P.; Tsinkalovsky, O.; Immervoll, H.; Bøe, S.O.; Svendsen, A.; Prestegarden, L.; Røsland, G.; Thorsen, F.; Stuhr, L.; et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int. J. Cancer 2008, 122, 761–768. [Google Scholar] [CrossRef]
- Brescia, P.; Richichi, C.; Pelicci, G. Current strategies for identification of glioma stem cells: Adequate or unsatisfactory? J. Oncol. 2012, 2012, 376894. [Google Scholar] [CrossRef]
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Anido, J.; Sáez-Borderías, A.; Gonzàlez-Juncà, A.; Rodón, L.; Folch, G.; Carmona, M.A.; Prieto-Sánchez, R.M.; Barba, I.; Martínez-Sáez, E.; Prudkin, L.; et al. TGF-β Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef]
- Lathia, J.D.; Gallagher, J.; Myers, J.T.; Li, M.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; Huang, A.Y.; Rich, J.N. Direct in vivo evidence for tumor propagation by glioblastoma cancer stem cells. PLoS ONE 2011, 6, e24807. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Li, Z.; Sathornsumetee, S.; Wang, H.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008, 68, 6043–6048. [Google Scholar] [CrossRef]
- Tchoghandjian, A.; Baeza, N.; Colin, C.; Cayre, M.; Metellus, P.; Beclin, C.; Ouafik, L.; Figarella-Branger, D. A2B5 cells from human glioblastoma have cancer stem cell properties. Brain Pathol. 2010, 20, 211–221. [Google Scholar] [CrossRef]
- Jin, X.; Kim, L.J.Y.; Wu, Q.; Wallace, L.C.; Prager, B.C.; Sanvoranart, T.; Gimple, R.C.; Wang, X.; Mack, S.C.; Miller, T.E.; et al. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat. Med. 2017, 23, 1352–1361. [Google Scholar] [CrossRef]
- Venere, M.; Fine, H.A.; Dirks, P.B.; Rich, J.N. Cancer stem cells in gliomas: Identifying and understanding the apex cell in cancer’s hierarchy. Glia 2011, 59, 1148–1154. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Laks, D.R.; Masterman-Smith, M.; Visnyei, K.; Angenieux, B.; Orozco, N.M.; Foran, I.; Yong, W.H.; Vinters, H.V.; Liau, L.M.; Lazareff, J.A.; et al. Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells 2009, 27, 980–987. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Rietze, R.L. Neural stem cells and neurospheres--re-evaluating the relationship. Nat. Methods 2005, 2, 333–336. [Google Scholar] [CrossRef]
- Pastrana, E.; Silva-Vargas, V.; Doetsch, F. Eyes wide open: A critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 2011, 8, 486–498. [Google Scholar] [CrossRef]
- den Hollander, P.; Joseph, R.; Vasaikar, S.; Kuburich, N.A.; Deshmukh, A.P.; Mani, S.A. Limiting Dilution Tumor Initiation Assay: An In Vivo Approach for the Study of Cancer Stem Cells. Methods Mol. Biol. 2022, 2429, 547–554. [Google Scholar]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef]
- Smith, L.R.; Cho, S.; Discher, D.E. Stem Cell Differentiation is Regulated by Extracellular Matrix Mechanics. Physiology 2018, 33, 16–25. [Google Scholar] [CrossRef]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef]
- Gangemi, R.M.; Griffero, F.; Marubbi, D.; Perera, M.; Capra, M.C.; Malatesta, P.; Ravetti, G.L.; Zona, G.L.; Daga, A.; Corte, G. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 2009, 27, 40–48. [Google Scholar] [CrossRef]
- Lathia, J.D.; Hitomi, M.; Gallagher, J.; Gadani, S.P.; Adkins, J.; Vasanji, A.; Liu, L.; Eyler, C.E.; Heddleston, J.M.; Wu, Q.; et al. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis. 2011, 2, e200. [Google Scholar] [CrossRef]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Stiles, C.D.; Rowitch, D.H. Glioma stem cells: A midterm exam. Neuron 2008, 58, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes. Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- Fouse SD, C.J. Cancer stem cells in brain tumors and their therapeutic implications. Neurotherapeutics 2009, 6, 694–702. [Google Scholar]
- Choi, J.D.; Lee, J.S. Interplay between Epigenetics and Genetics in Cancer. Genom. Inform. 2013, 11, 164–173. [Google Scholar] [CrossRef]
- Suvà, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef]
- Shah, A.H.; Rivas, S.R.; Doucet-O’Hare, T.T.; Govindarajan, V.; DeMarino, C.; Wang, T.; Ampie, L.; Zhang, Y.; Banasavadi-Siddegowda, Y.K.; Walbridge, S.; et al. Human endogenous retrovirus K contributes to a stem cell niche in glioblastoma. J. Clin. Investig. 2023, 133, 13. [Google Scholar] [CrossRef]
- Mehta, S.; Huillard, E.; Kesari, S.; Maire, C.L.; Golebiowski, D.; Harrington, E.P.; Alberta, J.A.; Kane, M.F.; Theisen, M.; Ligon, K.L.; et al. The central nervous system-restricted transcription factor Olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer Cell 2011, 19, 359–371. [Google Scholar] [CrossRef]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef]
- Yin, J.; Seo, Y.; Rhim, J.; Jin, X.; Kim, T.H.; Kim, S.S.; Hong, J.H.; Gwak, H.S.; Yoo, H.; Park, J.B.; et al. Cross-talk between PARN and EGFR-STAT3 Signaling Facilitates Self-Renewal and Proliferation of Glioblastoma Stem Cells. Cancer Res. 2023, 83, 3693–3709. [Google Scholar] [CrossRef]
- Pang, L.; Dunterman, M.; Guo, S.; Khan, F.; Liu, Y.; Taefi, E.; Bahrami, A.; Geula, C.; Hsu, W.H.; Horbinski, C.; et al. Kunitz-type protease inhibitor TFPI2 remodels stemness and immunosuppressive tumor microenvironment in glioblastoma. Nat. Immunol. 2023, 24, 1654–1670. [Google Scholar] [CrossRef] [PubMed]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, G.; Molaei, F.; Haider, S.; Almeida, M.P.; Lin, S.; Kushida, M.; Sureshkumar, H.; Bhatti, J.K.; Lu, J.Q.; Schramek, D.; et al. Fitness Screens Map State-Specific Glioblastoma Stem Cell Vulnerabilities. Cancer Res. 2024, 84, 3967–3983. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef]
- Folkins, C.; Shaked, Y.; Man, S.; Tang, T.; Lee, C.R.; Zhu, Z.; Hoffman, R.M.; Kerbel, R.S. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res. 2009, 69, 7243–7251. [Google Scholar] [CrossRef]
- Zhao, L.; Qiu, Z.; Yang, Z.; Xu, L.; Pearce, T.M.; Wu, Q.; Yang, K.; Li, F.; Saulnier, O.; Fei, F.; et al. Lymphatic endothelial-like cells promote glioblastoma stem cell growth through cytokine-driven cholesterol metabolism. Nat. Cancer 2024, 5, 147–166. [Google Scholar] [CrossRef]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, X.; Jin, K.; Zhu, J.; Wang, Y.; Xiong, S.; Mao, Y.; Zhou, L. B7-H4 is preferentially expressed in non-dividing brain tumor cells and in a subset of brain tumor stem-like cells. J. Neurooncol. 2008, 89, 121–129. [Google Scholar] [CrossRef]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.E.; Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar]
- Himes, S.R.; Sester, D.P.; Ravasi, T.; Cronau, S.L.; Sasmono, T.; Hume, D.A. The JNK are important for development and survival of macrophages. J. Immunol. 2006, 176, 2219–2228. [Google Scholar] [CrossRef]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Dean, M. ABC transporters, drug resistance, and cancer stem cells. J. Mammary Gland. Biol. Neoplasia 2009, 14, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef]
- Yuan, H.; Wu, X.; Wu, Q.; Chatoff, A.; Megill, E.; Gao, J.; Huang, T.; Duan, T.; Yang, K.; Jin, C.; et al. Lysine catabolism reprograms tumour immunity through histone crotonylation. Nature 2023, 617, 818–826. [Google Scholar] [CrossRef]
- Dahan, P.; Martinez Gala, J.; Delmas, C.; Monferran, S.; Malric, L.; Zentkowski, D.; Lubrano, V.; Toulas, C.; Cohen-Jonathan Moyal, E.; Lemarie, A. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: Possible involvement in radioresistance. Cell Death Dis. 2014, 5, e1543. [Google Scholar] [CrossRef]
- Lathia, J.D.; Li, M.; Hall, P.E.; Gallagher, J.; Hale, J.S.; Wu, Q.; Venere, M.; Levy, E.; Rani, M.R.; Huang, P.; et al. Laminin alpha 2 enables glioblastoma stem cell growth. Ann. Neurol. 2012, 72, 766–778. [Google Scholar] [CrossRef]
- Bidlingmaier, S.; Zhu, X.; Liu, B. The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells. J. Mol. Med. 2008, 86, 1025–1032. [Google Scholar] [CrossRef]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006, 12, 1167–1174. [Google Scholar] [CrossRef]
- Iudicone, P.; Fioravanti, D.; Cicchetti, E.; Zizzari, I.G.; Pandolfi, A.; Scocchera, R.; Fazzina, R.; Pierelli, L. Interleukin-15 enhances cytokine induced killer (CIK) cytotoxic potential against epithelial cancer cell lines via an innate pathway. Hum. Immunol. 2016, 77, 1239–1247. [Google Scholar] [CrossRef]
- Jackson, H.J.; Rafiq, S.; Brentjens, R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016, 13, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- City of Hope Medical Center A Phase 1 Study to Evaluate IL13Rα2-Targeted Chimeric Antigen Receptor (CAR) T Cells Combined with Checkpoint Inhibition for Patients with Resectable Recurrent Glioblastoma. Clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT04003649 (accessed on 19 October 2024).
- Wang, S.; Wei, W.; Yuan, Y.; Sun, B.; Yang, D.; Liu, N.; Zhao, X. Chimeric antigen receptor T cells targeting cell surface GRP78 efficiently kill glioblastoma and cancer stem cells. J. Transl. Med. 2023, 21, 493. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Gerstner, E.R.; Frigault, M.J.; Leick, M.B.; Mount, C.W.; Balaj, L.; Nikiforow, S.; Carter, B.S.; Curry, W.T.; Gallagher, K.; et al. Intraventricular CARv3-TEAM-E T Cells in Recurrent Glioblastoma. N. Engl. J. Med. 2024, 390, 1290–1298. [Google Scholar] [CrossRef]
- University of Florida. Phase I Study of IL-8 Receptor-modified CD70 CAR T Cell Therapy in CD70+ Adult Glioblastoma (IMPACT) (IMPACT) [Internet]. ClinicalTrials.gov. 2025. Available online: https://clinicaltrials.gov/study/NCT05353530 (accessed on 1 January 2025).
- City of Hope Medical Center. Chimeric Antigen Receptor (CAR) T Cells With a Chlorotoxin Tumor-Targeting Domain for the Treatment of MMP2+ Recurrent or Progressive Glioblastoma [Internet]. ClinicalTrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT04214392 (accessed on 9 November 2024).
- University of Pennsylvania. CART-EGFRvIII + Pembrolizumab in GBM [Internet]. ClinicalTrials.gov. 2023. Available online: https://clinicaltrials.gov/study/NCT03726515 (accessed on 22 June 2024).
- Bradshaw, A.; Wickremsekera, A.; Tan, S.T.; Peng, L.; Davis, P.F.; Itinteang, T. Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front. Surg. 2016, 3, 21. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Joo, K.M.; Kim, S.Y.; Jin, X.; Song, S.Y.; Kong, D.S.; Lee, J.I.; Jeon, J.W.; Kim, M.H.; Kang, B.G.; Jung, Y.; et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab. Investig. 2008, 88, 808–815. [Google Scholar] [CrossRef]
- Tamura, K.; Aoyagi, M.; Wakimoto, H.; Ando, N.; Nariai, T.; Yamamoto, M.; Ohno, K. Accumulation of CD133-positive glioma cells after high-dose irradiation by Gamma Knife surgery plus external beam radiation. J. Neurosurg. 2010, 113, 310–318. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Hamburg, E.J.; Atit, R.P. Sustained β-catenin activity in dermal fibroblasts is sufficient for skin fibrosis. J. Investig. Dermatol. 2012, 132, 2469–2472. [Google Scholar] [CrossRef]
- Bhat, K.P.; Salazar, K.L.; Balasubramaniyan, V.; Wani, K.; Heathcock, L.; Hollingsworth, F.; James, J.D.; Gumin, J.; Diefes, K.L.; Kim, S.H.; et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes. Dev. 2011, 25, 2594–2609. [Google Scholar] [CrossRef]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert. Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef]
- Heinrich, M.A.; Bansal, R.; Lammers, T.; Zhang, Y.S.; Michel Schiffelers, R.; Prakash, J. 3D-Bioprinted Mini-Brain: A Glioblastoma Model to Study Cellular Interactions and Therapeutics. Adv. Mater. 2019, 31, e1806590. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef]
- Navin, N.E. Cancer genomics: One cell at a time. Genome Biol. 2014, 15, 452. [Google Scholar] [CrossRef]
- Tirosh, I.; Suvà, M.L. Tackling the Many Facets of Glioblastoma Heterogeneity. Cell Stem Cell 2020, 26, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef] [PubMed]
- Hood, L.; Friend, S.H. Predictive, personalized, preventive, participatory (P4) cancer medicine. Nat. Rev. Clin. Oncol. 2011, 8, 184–187. [Google Scholar] [CrossRef]
- Schilsky, R.L. Personalized medicine in oncology: The future is now. Nat. Rev. Drug Discov. 2010, 9, 363–366. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Rizvanov, A.A.; Solovyeva, V.V. The Role of Cancer Stem Cells and Their Extracellular Vesicles in the Modulation of the Antitumor Immunity. Int. J. Mol. Sci. 2022, 24, 395. [Google Scholar] [CrossRef]
- Dean, M. Cancer stem cells: Redefining the paradigm of cancer treatment strategies. Mol. Interv. 2006, 6, 140–148. [Google Scholar] [CrossRef]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes. Dev. 2012, 26, 756–784. [Google Scholar] [CrossRef]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef]
- Audia, A.; Conroy, S.; Glass, R.; Bhat, K.P.L. The Impact of the Tumor Microenvironment on the Properties of Glioma Stem-Like Cells. Front. Oncol. 2017, 7, 143. [Google Scholar] [CrossRef]
- Al Khzem, A.H.; Gomaa, M.S.; Alturki, M.S.; Tawfeeq, N.; Sarafroz, M.; Alonaizi, S.M.; Al Faran, A.; Alrumaihi, L.A.; Alansari, F.A.; Alghamdi, A.A. Drug Repurposing for Cancer Treatment: A Comprehensive Review. Int. J. Mol. Sci. 2024, 25, 12441. [Google Scholar] [CrossRef] [PubMed]
- Prados, M.D.; Byron, S.A.; Tran, N.L.; Phillips, J.J.; Molinaro, A.M.; Ligon, K.L.; Wen, P.Y.; Kuhn, J.G.; Mellinghoff, I.K.; De Groot, J.F.; et al. Toward precision medicine in glioblastoma: The promise and the challenges. Neuro Oncol. 2015, 17, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs. Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef]

{kind=link}
| Marker | Expression/Localization | Key Functional Role | Relevance in GSC Biology | Potential for Targeted Therapy | Additional Comments |
|---|---|---|---|---|---|
| CD133 | Cell-surface glycoprotein (also known as Prominin-1) | Maintenance of stem-like phenotype; Self-renewal | Widely used to isolate GSCs; Expression correlates with poor prognosis | Potential immunotherapy target (e.g., vaccines, antibodies); Strategies under investigation |
|
| CD44 | Cell-surface adhesion molecule | Cell adhesion and migration; Contributes to mesenchymal/invasive properties | Enriched in mesenchymal GSC subtypes; Facilitates brain infiltration | Blockade strategies (e.g., antibodies, small-molecule inhibitors) explored |
|
| A2B5 | Cell-surface ganglioside marker | Identifies glial precursor-like cells | Widely co-expressed with other stem markers (e.g., CD133); Helps refine GSC populations | Possible immunotherapeutic target in combination with other markers |
|
| L1CAM | Cell-surface adhesion molecule | Promotes cell motility and adhesion; Enhances invasiveness | Crucial for GSC maintenance and survival; Associated with radiation resistance | Monoclonal antibodies under development |
|
| Integrin α6 | Cell-surface receptor for laminin | Mediates cell-ECM attachment; Promotes survival and invasion | High levels correlate with stemness; Facilitates basement membrane infiltration | Integrin inhibitors in clinical or preclinical evaluation |
|
| Nestin | Intracellular intermediate filament protein | Structural support in progenitor cells | Neural stem/progenitor cell marker; Reflects high proliferative capacity | Not directly targeted; Primarily used for GSC identification |
|
| SOX2 | Intracellular transcription factor | Maintains pluripotency and self-renewal | Essential for GSC proliferation; Drives stem-like gene programs | Various small-molecule inhibitors under early investigation |
|
| OLIG2 | Intracellular transcription factor | Regulates oligodendrocyte lineage commitment; Contributes to neuronal specification | Critical for GSC proliferation; Associated with radioresistance | Potential gene therapy or epigenetic modulation |
|
| BMI1 | Intracellular polycomb group protein | Chromatin remodeling; Governs self-renewal | Promotes GSC survival; Linked to therapy resistance and aggressiveness | Epigenetic inhibitors targeting BMI1 are being tested |
|
| ALDH1A3 | Cytoplasmic enzyme (aldehyde dehydrogenase) | Detoxification; Retinoic acid metabolism | Enriched in tumor-initiating GSC subpopulations; Associated with chemo- and radioresistance | ALDH inhibitors show promise in preclinical models |
|
| PDLIM1 | Intracellular scaffold protein containing PDZ and LIM domains; also known as CLP36 | Regulates proliferation, apoptosis, and tumorigenesis; Maintains/expands GSC subpopulations; Confers chemoresistance (via PI3K-AKT pathway) | Specifically enriched in GSCs within GBM; Drives poor prognosis and therapy resistance | Novel target for inhibiting GSC-mediated tumor growth and resistance |
|
| GSC Subtype | Developmental | Injury-Response |
|---|---|---|
| Key Transcriptional Programs and Features |
|
|
| Unique Dependencies/Vulnerabilities |
|
|
| Targeted Pathways/Genes |
|
|
| Representative Experimental Drugs or Approaches |
|
|
| Rationale/Mechanism |
|
|
| Mechanism | Important Molecules | GSC-Driven Effects | Potential Interventions |
|---|---|---|---|
| Secretion of immunosuppressive cytokines |
|
|
|
| Immune checkpoint molecule expression |
|
|
|
| GSC-modulated APC dysfunction |
|
|
|
| Rewiring of amino acid metabolism |
|
|
|
| Enhanced extracellular stress response |
|
|
|
| Combination Strategy | Mechanism of Action | Synergy and Key Findings | Evidence (Preclinical/Clinical) |
|---|---|---|---|
| GSC-Targeted Inhibitors + SOC (TMZ/Radiotherapy) |
|
| Preclinical and early-phase trials |
| Small-Molecule EGFR Inhibitors (e.g., Erlotinib) + SOC |
|
| Preclinical studies |
| Immune Checkpoint Inhibitors + GSC-Targeted Therapy |
|
| Ongoing clinical trials |
| CAR T Cells + Standard-of-Care |
|
| Phase 1/2 clinical trials |
| Metabolic Inhibitors (e.g., Lysine Restriction) + Immune Tx |
|
| Preclinical models |
| Angiogenesis Inhibitors (Bevacizumab) + GSC-Directed Agents |
|
| Preclinical and clinical settings |
| Epigenetic Modulators (HDAC/BMI1 Inhibitors) + SOC |
|
| Preclinical |
| Multi-Targeted Approach: GSC-Targeted Vaccine + Checkpoint Inhibitors + SOC |
|
| Early clinical trials |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.; Amin, M.A.; Campian, J.L. Glioblastoma Stem Cells at the Nexus of Tumor Heterogeneity, Immune Evasion, and Therapeutic Resistance. Cells 2025, 14, 562. https://doi.org/10.3390/cells14080562
Tang J, Amin MA, Campian JL. Glioblastoma Stem Cells at the Nexus of Tumor Heterogeneity, Immune Evasion, and Therapeutic Resistance. Cells. 2025; 14(8):562. https://doi.org/10.3390/cells14080562
Chicago/Turabian StyleTang, Justin, Md Al Amin, and Jian L. Campian. 2025. "Glioblastoma Stem Cells at the Nexus of Tumor Heterogeneity, Immune Evasion, and Therapeutic Resistance" Cells 14, no. 8: 562. https://doi.org/10.3390/cells14080562
APA StyleTang, J., Amin, M. A., & Campian, J. L. (2025). Glioblastoma Stem Cells at the Nexus of Tumor Heterogeneity, Immune Evasion, and Therapeutic Resistance. Cells, 14(8), 562. https://doi.org/10.3390/cells14080562