Genes 2022, 13(12), 2372; https://doi.org/10.3390/genes13122372 - 15 Dec 2022
Cited by 5 | Viewed by 1447
Abstract
Gentiana macrophylla Pall. (G. macrophylla)—a member of the family Gentianaceae—is a well-known traditional Chinese medical herb. Iridoids are the main active components of G. macrophylla, which has a wide range of pharmacological activities such as dispelling wind, eliminating dampness, clearing
[...] Read more.
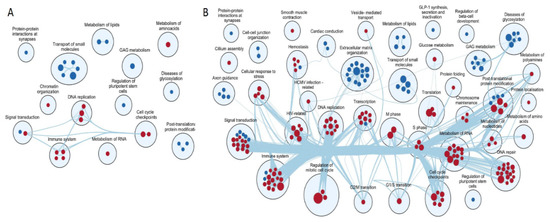
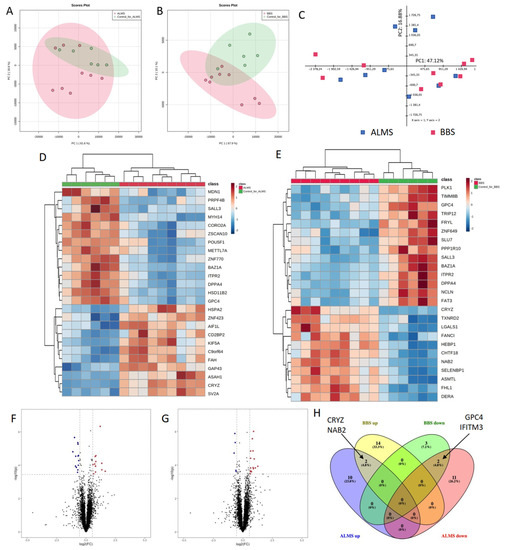
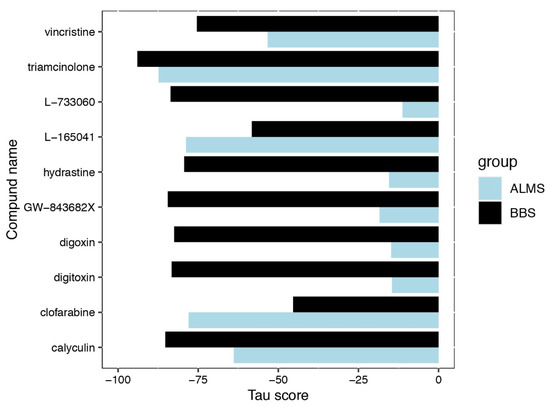
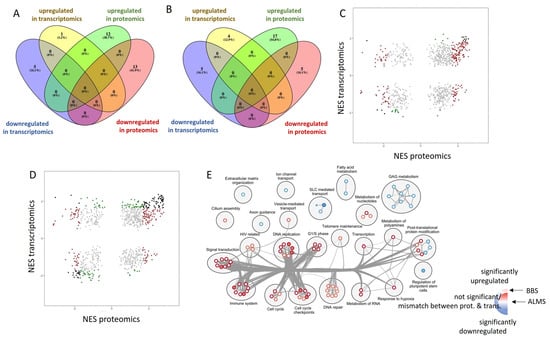
Gentiana macrophylla Pall. (G. macrophylla)—a member of the family Gentianaceae—is a well-known traditional Chinese medical herb. Iridoids are the main active components of G. macrophylla, which has a wide range of pharmacological activities such as dispelling wind, eliminating dampness, clearing heat and asthenic fever, hepatoprotective and choleretic actions, and other medicinal effects. In this study, a total of 67,048 unigenes were obtained by transcriptomic sequencing analysis of G. macrophylla. A BLAST analysis showed that 48.21%, 33.66%, 46.32%, and 32.62% of unigenes were identified in the NR, Swiss-Prot, eggNOG, and KEGG databases, respectively. Twenty-five key enzymes were identified in the iridoid biosynthesis pathway. Most of the upregulated unigenes were enriched in flowers and leaves. The trustworthiness of the transcriptomic data was validated by real-time quantitative PCR (qRT-PCR). A total of 22 chemical constituents were identified by ultra-high performance liquid chromatography-quadrupole-electrostatic field Orbitrap mass spectrometry (UPLC-Q-Exactive MS), including 10 iridoids. A correlation analysis showed that the expression of 7-DLH and SLS was closely related to iridoids. The expression of 7-DLH and SLS was higher in flowers, indicating that flowers are important for iridoid biosynthesis in G. macrophylla.
Full article
(This article belongs to the Special Issue Genetic Mechanism of Plant Responses to Environmental Stresses)
►
Show Figures

Figure 1








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}