Abstract
The Rhus gall aphid Schlechtendalia chinensis uses the species Rhus chinensis as its primary host plant, on which galls are produced. The galls have medicinal properties and can be used in various situations due to their high tannin content. Detoxification enzymes play significant roles in the insect lifecycle. In this study, we focused on five detoxification gene families, i.e., glutathione-S-transferase (GST), ABC transporter (ABC), Carboxylesterase (CCE), cyto-chrome P450 (CYP), and UDP-glycosyltransferase (UDP), and manually annotated 144 detoxification genes of S. chinensis using genome-wide techniques. The detoxification genes appeared mostly on chromosome 1, where a total of two pair genes were identified to show tandem duplications. There were 38 gene pairs between genomes of S. chinensis and Acyrthosiphon pisum in the detoxification gene families by collinear comparison. Ka/Ks ratios showed that detoxification genes of S. chinensis were mainly affected by purification selection during evolution. The gene expression numbers of P450s and ABCs by transcriptome sequencing data were greater, while gene expression of CCEs was the highest, suggesting they might be important in the detoxification process. Our study has firstly identified the genes of the different detoxification gene families in the S. chinensis genome, and then analyzed their general features and expression, demonstrating the importance of the detoxification genes in the aphid and providing new information for further research.
1. Introduction
The Rhus gall (or sumac-gall) aphids switch host plants between Rhus species and mosses to finish their life cycles, and form galls on their primary host plants Rhus (Anacardiaceae) species [1,2,3]. The galls are often known as the Chinese galls and they are rich in tannins and economically important in Asia because galls have medicinal properties and represent sources of industrial tannin [4,5]. This aphid group belonged to the subtribe Melaphidina of tribe Fordini (Aphididae: Eriosomatinae) [6,7,8], including six genera and 12 species [3], among which S. chinensis is the most common and wide-spread species, with R. chinensis as its unique primary host plant and Mniaceae species as its secondary hosts, as well as a life cycle including both sexual and asexual reproduction stages [9,10].
Phytophagous insects and host plants have coevolved, and plants have evolved physical or chemical defense mechanisms to resist insect feeding, while insects have evolved perfect anti-defense mechanisms [11]. Insects adapted to host plants partly rely on their detoxification genes, whose mechanism has been divided into three stages, i.e., an initial oxidation/reduction-hydrolysis involving mainly cytochrome P450 monooxygenase (P450) and carboxylesterases (CCE), enzymatic conjugation UDP-glucuronyl transferases (UGTs) or glutathione S-transferases (GST), and conjugated-metabolite transport-excretion out of the cells (ABC transporters (ABC)) [12,13,14].
The P450 gene family (P450s) has the ability to diminish the biological activity of a wide range of endogenous toxic compounds and exogenous substances [15]. Thomas et al. (2021) annotated 66 cytochrome P450s in Phylloxera Daktulosphaira vitifoliae and classified them into four clades [16]. Carboxylesterase (CCEs) belongs to one gene family of the α/β hydrolase protein superfamily [17], and fifty-seven putative CCEs was identified in Anopheles sinensis and divided into three classes, 12 subfamilies and 14 clades [18]. Glutathione S-transferases (GSTs) and uridine diphosphate-glycosyltransferases (UGTs) are conjugation enzymes, that covalently attach small endogenous hydrophilic molecules in order to increase compound solubility and facilitate their excretion, which have effects on the toxic by-products of phase I metabolism [19]. Thirty-six putative cytosolic GSTs and five microsomal GSTs were identified in Tribolium castaneum to reveal the largest insect-specific GSTs: Epsilon and Delta [20]. ATP-binding cassette transporters (ABC) transport substrates including amino acids, lipids, peptides, sugars and drugs across cell membranes by using ATP hydrolysis energy [21]. A total of 47 ABC genes in Bactrocera dorsalis were identified and classified into eight subfamilies (A–H), and it was suggested that these genes may play important roles in xenobiotic metabolism and biosynthesis in B. dorsalis [22].
However, there is no report on the detoxification genes in the Rhus gall aphid S. chinensis genome. Here, we used the third-generation sequencing technology to obtain the whole genome of S. chinensis at the chromosome level and performed the comprehensive analysis of five detoxification gene families in the S. chinensis genome. In detail, we conducted systematic identification and molecular characterization, which included gene family member identification, collinear analysis, chromosomal location, Ka/Ks evolutionary selection pressure analysis, gene expression analysis, protein physicochemical properties, and structure prediction. We highlight the characters of the key detoxification genes in S. chinensis-R. chinensis adaptive interactions for future functional studies.
2. Materials and Methods
2.1. Sample Information
Mature and fresh Rhus galls were collected from the garden in Shanxi University, Shanxi, China. The gall was cut open and live aphids were used for third-Generation Sequencing. In addition, we selected the aphid samples from three mature galls, numbered A4601, A4603, and A4621 for transcription sequencing to characterize the gene expression pattern of detoxification genes in S. chinensis. The specimens were stored at the School of Life Science, Shanxi University, China.
2.2. Identification of Detoxification Genes from S. chinensis
We sequenced the whole genome of S. chinensis by third—generation sequencing on the Pacbio platform. The protein-coding genes in the genome were annotated by integrating three approaches, namely de novo prediction, homology search, and transcript-based assembly. The protein sequences of the detoxification genes were obtained by searching in the annotation table of S. chinensis using key words of detoxification genes, and then confirmed in the NCBI Conserved Domain Database (NCBI-CDD) (e-value = 1 × 10−3), excluding those lacking conserved domains from the analysis [23].
2.3. Phylogenetic Tree, Motif Pattern, Domain, Gene Structure of Detoxification Genes
The protein coding sequences of detoxification genes of the three aphid species, A. pisum, Cinara cedri and Myzus persicae, were downloaded from the Insect BASE website (http://v2.insect-genome.com/, accessed on 12 May 2022). We performed the multiple-sequence alignment of detoxification genes using ClustalW software (created by Kumar et al.; Philadelphia, PA, USA) [24]. The alignment results were exported to fasta format, and then opened using tbtools to trim using Trimmer [25]. The protein sequences with large differences were filtered out. We constructed a phylogenetic tree using the Neighbor-Joining (NJ) method with the parameters of Poisson model, complete deletion and 1000 bootstrap replicates, and visualized and improved the tree using the program Evolview (http://www.evolgenius.info/evolview/, accessed on 18 March 2022) [26]. The relative frequency of the corresponding amino acid at each position was calculated on the Web Logo online website (http://weblogo.threeplusone.com/, accessed on 18 March 2022). We conducted motif analysis of the detoxification genes on the MEME online server (http://meme-suite.org/tools/meme, accessed on 20 March 2022) with parameters “minimum width = 6, maximum width = 50, number of motif to find = 10” [27], and the analysis of conserve domain by the NCBI Conserved Domain Database (NCBI-CDD) (e-value = 1 × 10−3) [23]. The exon and intron structures were displayed in all protein sequences using the Gene Structure Display Server (GSDS) (http://gsds.cbi.pku.edu.cn/, accessed on 22 March 2022) [28]. Finally, the corresponding results were visualized by the program TBtools [29].
2.4. Chromosomal Locations, Collinearity and Selection Pressure
The detoxification genes’ positions on chromosomes were displayed using TBtools (version 1.09876, created by Chen et al.; Wuhan, China) [29]. We conducted and visualized collinearity analysis by MCScanX (created by Wang et al.; Wuhan, China) [30] and Circos (version 2.50, created by Krzywinski et al., Vancouver, Canada) [31]. In addition, we used Ka Ks_Calculator 2.0 (version 2.0, created by Zhang et al.; Beijing, China) to calculate the ratio between the non-synonymous replacement rate (Ka) and the synonymous replacement rate (Ks) of two protein-coding genes, which is an indicator of molecular evolution to determine whether there is selective pressure on a protein-coding gene [32].
2.5. Expression Profile of Detoxification Genes
We extracted total mixed RNA from the three S. chinensis samples by the Trizol method [33], and constructed a sequence library using the Illumina TruseqTM RNA sample prep Kit [34]. On the Illumina HiSeq2500 platform, we carried out next generation sequencing for the sequence library. We measured the original raw reads and removed the low-quality, repetitive data with adapters to obtain clean reads. Finally, we assembled the transcriptome data from scratch to obtain the unigenes sequence by the Trinity assembly software [35]. Meanwhile, we compared unigenes sequence with six major databases, i.e., NR, Swiss-Prot, Pfam, COG, GO, and KEGG, to complete functional annotation and classification analysis [36,37,38,39,40,41,42,43]. We screened detoxification-related genes with our gene annotation lists. We compared the CDS sequences of the genomic detoxification genes with the transcriptome detoxification gene by MAFFT Alignment to determine the gene expression [44].
2.6. Prediction of Characteristics and Physicochemical Properties of Detoxification Gene
We used the online bioinformatics software Expasy Protoparam (https://web.expasy.org/protparam/, accessed on 23 March 2022) to predict the protein length, molecular weight, and isoelectric point of the keratins [45], and Signal P 5.0 Server (http://www.cbs.dtu.dk/services/SignalP/index.php, accessed on 24 March 2022) and TMHMM Server v.2.0 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0, accessed on 24 March 2022) to predict signal peptides, respectively [46,47].
2.7. Protein Structure Prediction from the Detoxification Genes
We analyzed the protein secondary structures of the detoxification gene products of S. chinensis by the program SOPMA with the website (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html, accessed on 24 March 2022) [48]. We predicted the tertiary structure of S. chinensis by Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index, accessed on 24 March 2022) [49].
3. Results
3.1. Identification of Detoxification Genes of S. chinensis
We identified nine genes in GSTs, 55 genes in ABCs, 18 genes in CCEs, 48 genes in CYPs and 14 genes in UDPs in the S. chinensis genome (Table 1). These genes were divided into three subfamilies for GSTs, i.e., delta, theta, and sigma, six subfamilies for ABCs, i.e., A, B, C, D, E, F, and G, four subfamilies for P450s, i.e., CYP2, CYP6, CYP4, and mitochondrion, two subfamilies UGT4 and UGT5 for UDPs. Detoxification gene numbers displayed heterogeneity in the annotated Aphidinae genomes. For example, A. pisum displayed more genes of GSTs than other aphid species due to having more genes in delta subfamily. S. chinensis had the greatest numbers in ABCs due to more genes in the C subfamily, and fewer in CCEs and CYPs due to fewer genes in Esterase and CYP6 subfamilies, respectively. These gene families, i.e., GSTs, ABCs, and CCEs, clearly exhibited expansion and contraction.

Table 1.
Comparison of detoxification gene numbers in annotated insect species.
3.2. Characteristic of the Five Detoxification Genes of S. chinensis
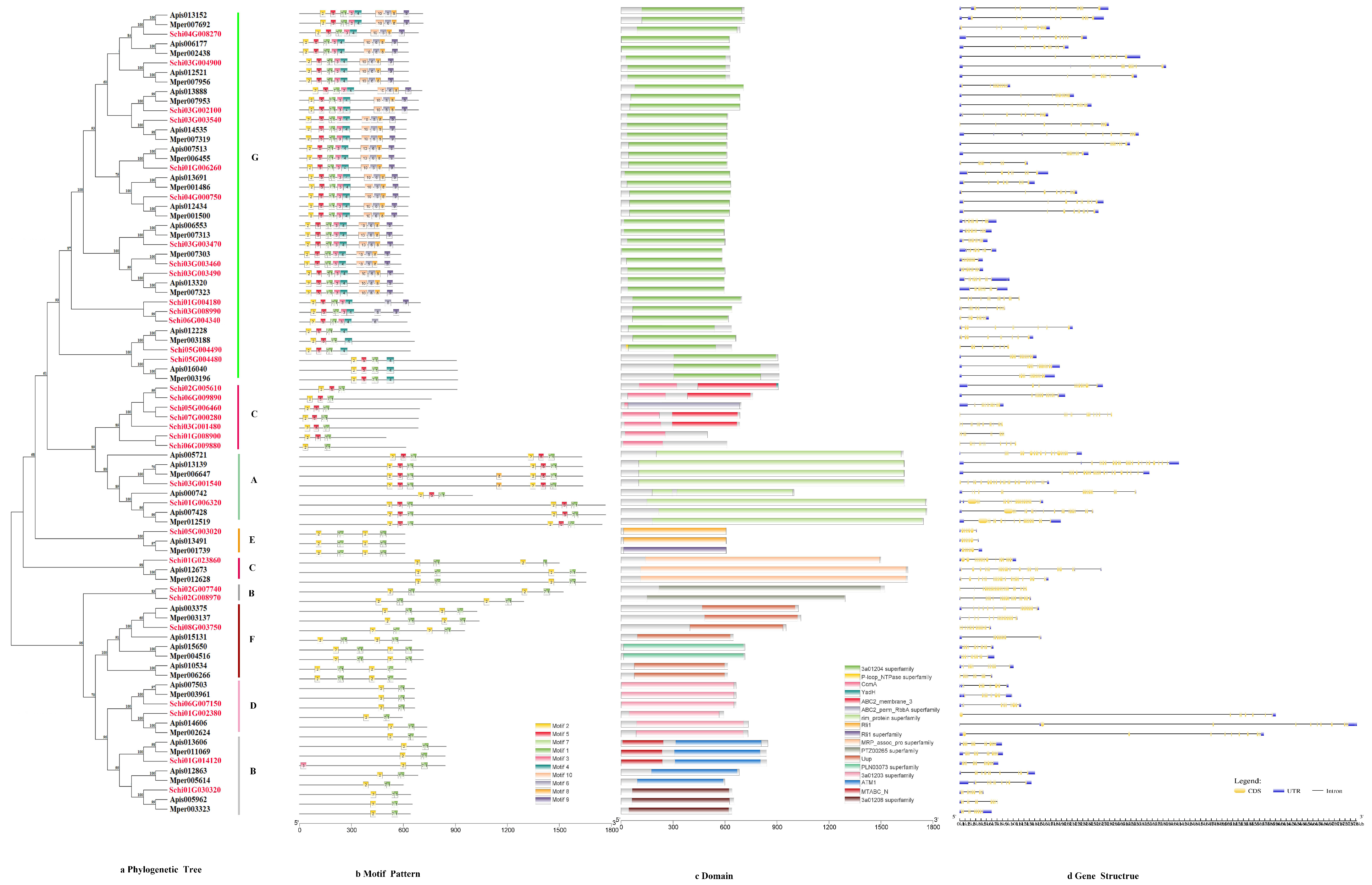
We performed a characteristic analysis of the detoxification genes, including motif, domain, and the number of exons, and constructed the phylogenetic tree of the protein sequences of five detoxification gene families from the four aphid species S. chinensis, A. pisum, C. cedri, and M. persicae.
3.2.1. P450s
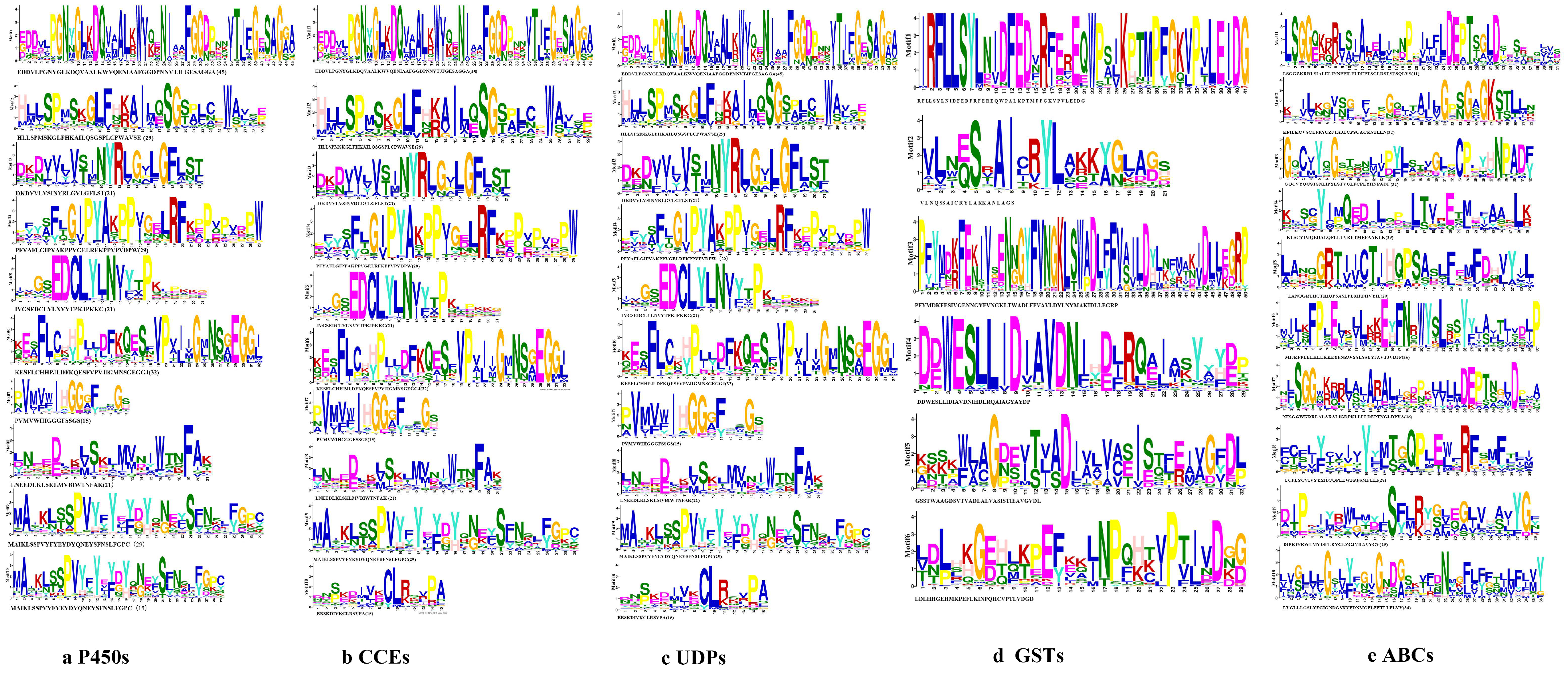
The phylogenetic tree of the P450s divided the sequences into four subfamilies, i.e., CYP4, CYP2, mitochondrial clan and CYP6 (Figure 1a). The genes in the same class had similar motif patterns and domain. For example, most of the motif order of S. chinensis in the CYP4 class were 10-6-8-5-4-3-2-1-7, except for Schi08G001700 with 6-3. All the CYP2 class motif was 5-4-3-2-1-7. The mitochondrial clan motif order was different with 4-3-2-1, 5-4-3-2-1 or 6-5-4-3-2-1-7. The CYP6 class motif order was 9-6-8-5-4-3-2-1-7 (Figure 1b). The length of ten conserved motifs of P450s varied from 15 to 50 amino acids (Figure 2a). The conserved domain included CYP4, cytochrome_P450, AdoMet_MTases, CYP24A1, CYP1_2, CYP15A1 and CYP6 (Figure 1c). The numbers of exons ranged from three to 22 from predictions of the gene structure (Figure 1d). Gene length varied from 0 to 61kb in the P450s, among which the majority (65%) were 0–10 kb, and a small proportion (35%) of the genes were greater than 10 kb in size.
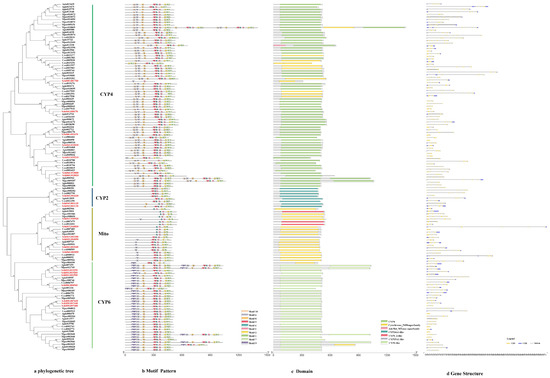
Figure 1.
(a) Phylogenetic relationships, (b) conserved motifs, (c) domains and (d) gene structure analysis of detoxification genes of the P450 gene family in S. chinensis, A. pisum, C. cedri, and M. persicae.

Figure 2.
The 10 conserved motifs of detoxification genes families in the S. chinensis. (a) P450 gene family. (b) CCE gene family. (c) UDP gene family. (d) GST gene family. (e) ABC gene family.
3.2.2. CCEs
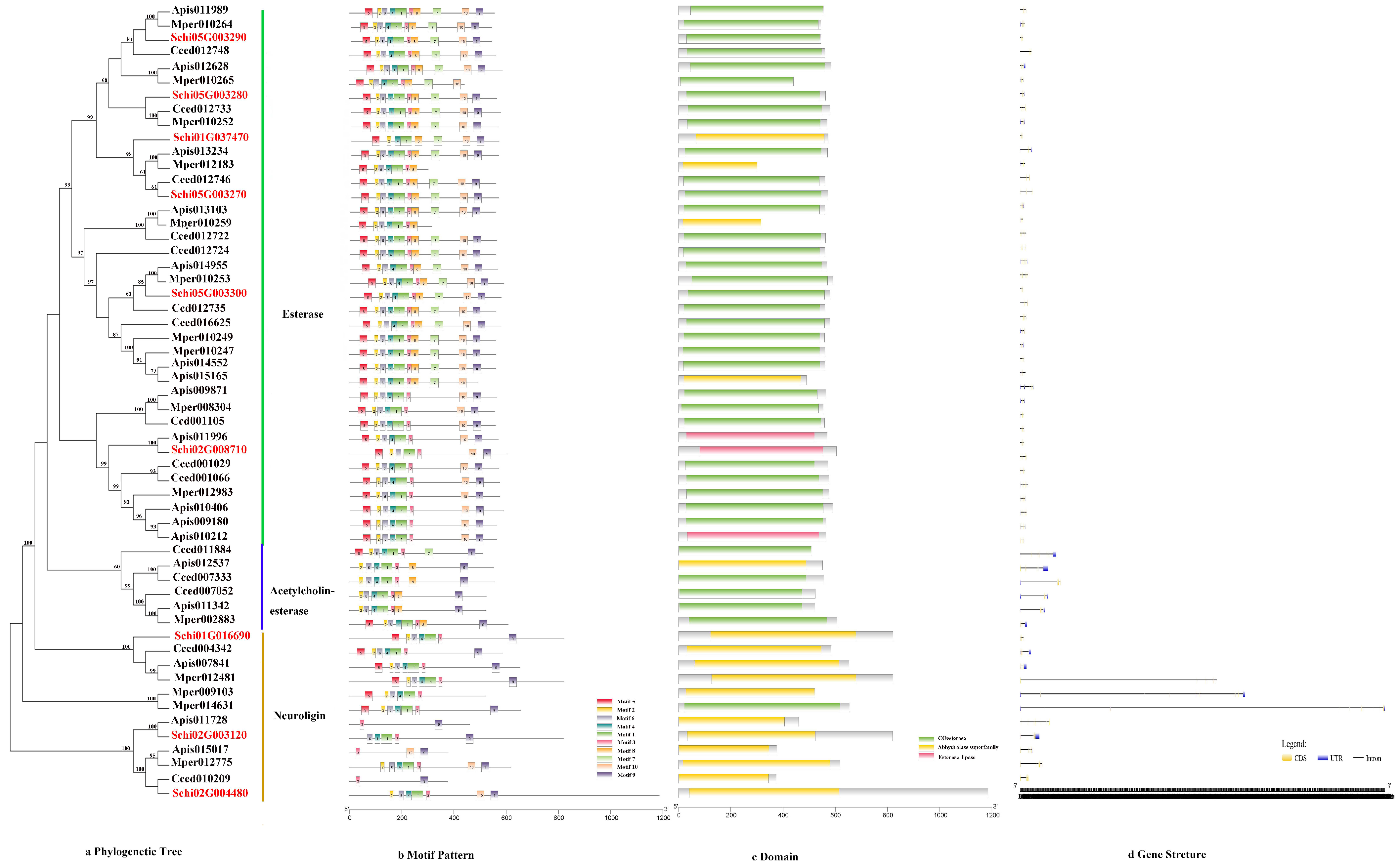
The phylogenetic tree of the CCEs divided the detoxification genes into three subfamilies, i.e., Esterase, Acetylcholinesterase and Neuroligin (Figure 3a). The motif order of the three classes was different, but most of them had the common order 2-6-4-1-3-8-7-10-9 (Figure 3b). Ten conserved motifs of CCEs varied from 15 to 45 amino acids in length (Figure 2b). The conserved domain of CCEs included coesterase, Abhydrolase and esterase _lipase (Figure 3c). The analysis on the coding sequence (CDS) and untranslated regions (UTRs) of the CCEs showed that exon numbers ranged from two to 18, and a total of 41 members exhibited 5′and 3′ UTRs, eight members presented no UTR and the remaining seven members had either a 5′or 3′ UTR (Figure 3d). Differences in the motif pattern and gene structure of the different classes might be the reason for the differences in their physiological functions.
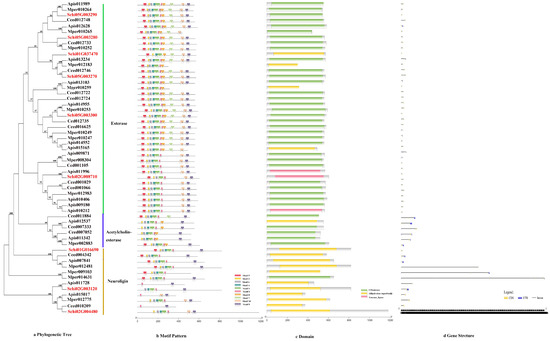
Figure 3.
(a) Phylogenetic relationships, (b) conserved motifs, (c) domains and (d) gene structures of detoxification genes of the CCE gene family in S. chinensis, A. pisum, C. cedri, and M. persicae.
3.2.3. UDPs
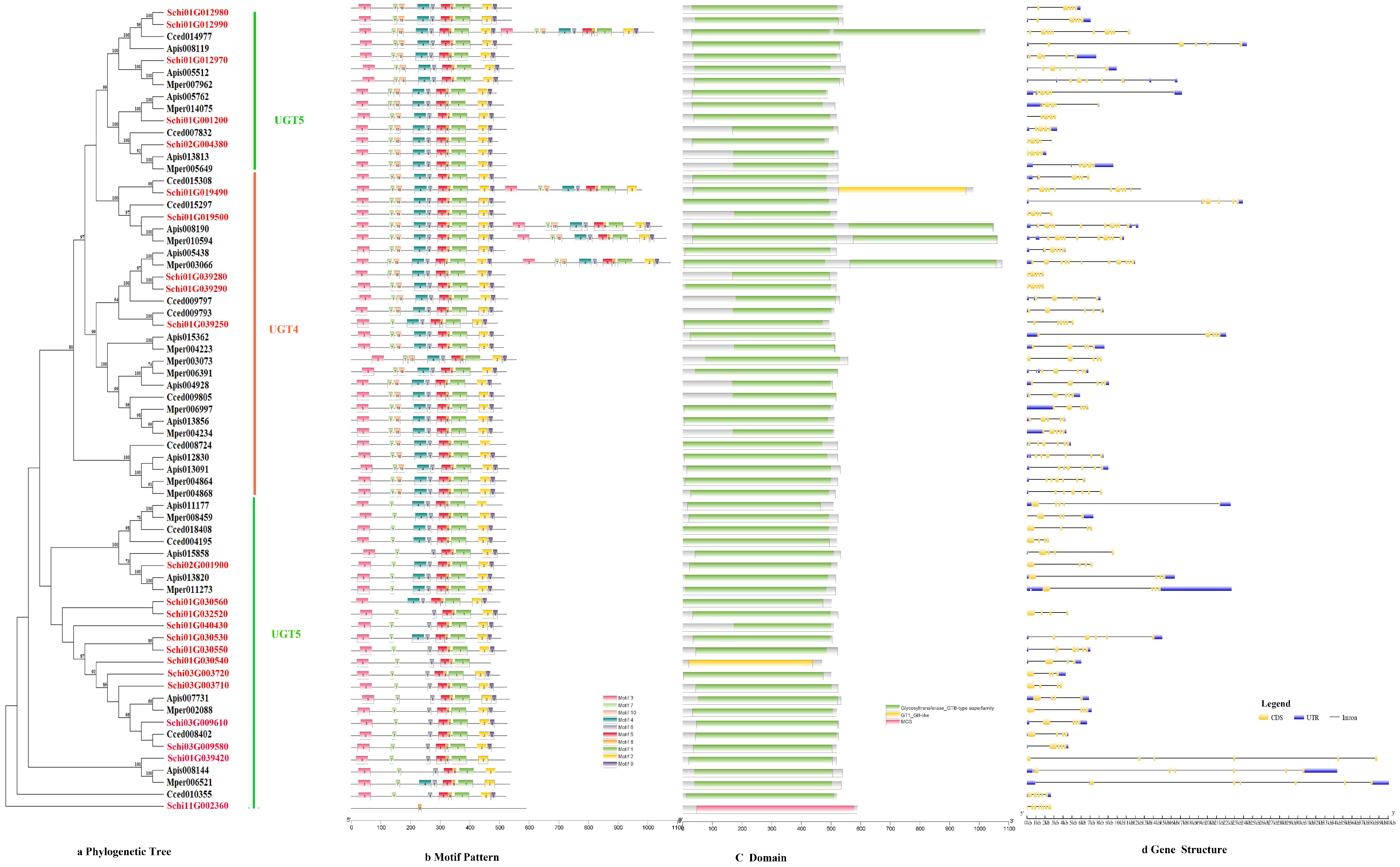
The phylogenetic tree divided the protein sequence of UDPs into two subfamilies, i.e., UGT4 and UGT5 (Figure 4a). All genes of UDPs showed the same motif order 3-7-10-4-6-5-8-1-2-9, only few genes lacked a 1-2 motif, i.e., Schi02G001900, Schi01G030560 (Figure 4b). Ten conserved motifs of UDPs varied from 15 to 50 amino acids in length (Figure 2c). The conserved domain of UDPs included Glycosyltransferase _GTB, GT1_Gtf and MCS (Figure 4c). Analysis of the coding sequence (CDS) and untranslated regions (UTRs) of the UDPs showed that the numbers of exons ranged from three to 11. Only two genes had 11 exons, 27 UDP genes (17.3%) had five exons, and 25 UDP genes (81.7%) had four exons. A total of 43 members exhibited 5′and 3′ UTRs, 13 members presented no UTR, and the remaining five members had either 5′or 3′UTR. The result shows that the same class has the same number of exons in the UDP gene family (Figure 4d).
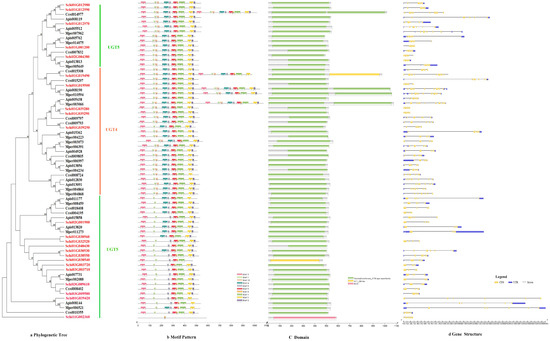
Figure 4.
(a) Phylogenetic relationships, (b) conserved motifs, (c) domains and (d) gene structures of detoxification genes of the UDP gene family in S. chinensis, A. pisum, C. cedri, and M. persicae.
3.2.4. GSTs
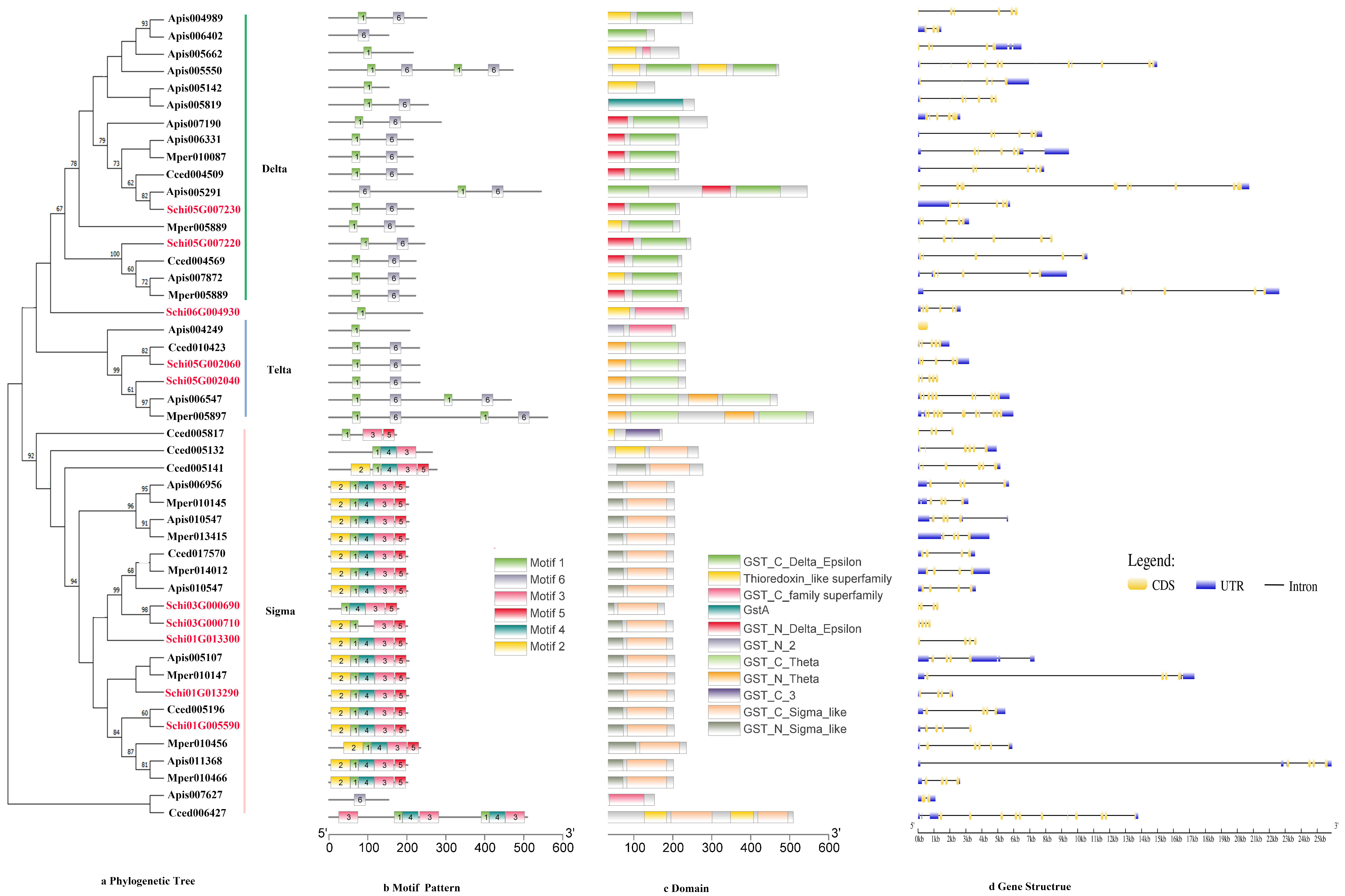
The phylogenetic tree divided the protein sequence of GSTs into three class, i.e., Delta, Theta and Sigma (Figure 5a). The delta subfamily included four genes, i.e., Schi05G007220, Schi05G002040, Schi05G002060, and Schi05G007230, which shared the same motif order (1-6) as the nine genes in the Sigma subfamily. Schi01G005590, Schi01G013290, Schi01G013300, Schi03G000690 and Schi03G000710 belonged to theta subfamily and shared the same motif order (3-1-4-2-5) (Figure 5b). Details of six putative motifs are outlined in Figure 2d. These conserved motifs ranged from 15 to 50 amino acids in length. The conserved domain of GSTs included GST _ Delta _ Epsilon, Gst A, GST _Theta, GST _Sigma and Thioredoxin (Figure 5c). In the delta and theta subfamily, the numbers of most exons were four, a few exons were just one. In the sigma subfamily, the numbers of most exons were 11, and other members had five exons (Figure 5d). The genes in the same groups had similar motif patterns and numbers of exons, indicating that they were highly conserved and the inferred functions were similar.
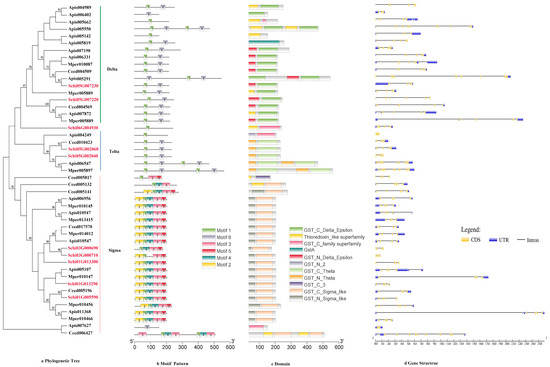
Figure 5.
(a) Phylogenetic relationships, (b) conserved motifs, (c) domains and (d) gene structures of detoxification genes of the GST gene family in S. chinensis, A. pisum, C. cedri, and M. persicae.
3.2.5. ABCs
The phylogenetic tree of the protein sequences of ABCs of S. chinensis were distributed to G, C, A, E, C, B, F and D subfamilies (Figure 6a). The motif order for G class was 2-5-7-1-3-4-10-6-8-9. The C class motif order was 2-5-7-1. A class motif order for most members was 2-5-7-1, with a few members (Schi03G001540) having special motif 8. The E, C, B, F and D class motif order was 2-7-1 (Figure 6b). The lengths of ABC conserved motifs ranged from 28 to 41 amino acids (Figure 2e). The conserved domain of ABCs included 3a1204, CcmA, YadH, Rli1, Uup, ATM1, MTABC and ABC2_membrane_3 (Figure 6c). The numbers of exon ranged from six to 28 by the predictions of the gene structure. The exon number of most members was 14-28. Analysis of the coding sequence (CDS) and untranslated regions (UTRs) of the ABC gene family found that just six members did not have a UTR (Figure 6d).
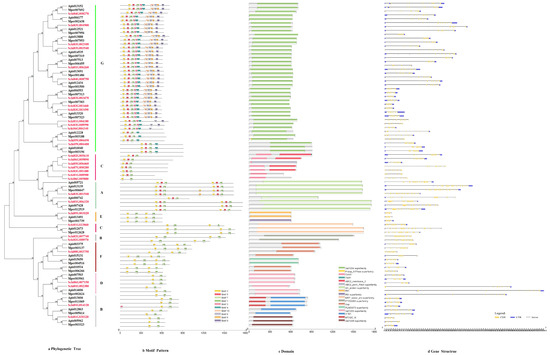
Figure 6.
(a) Phylogenetic relationships, (b) conserved motifs, (c) domains and (d) gene structures of detoxification genes of the ABC gene family in S. chinensis, A. pisum, C. cedri, and M. persicae.
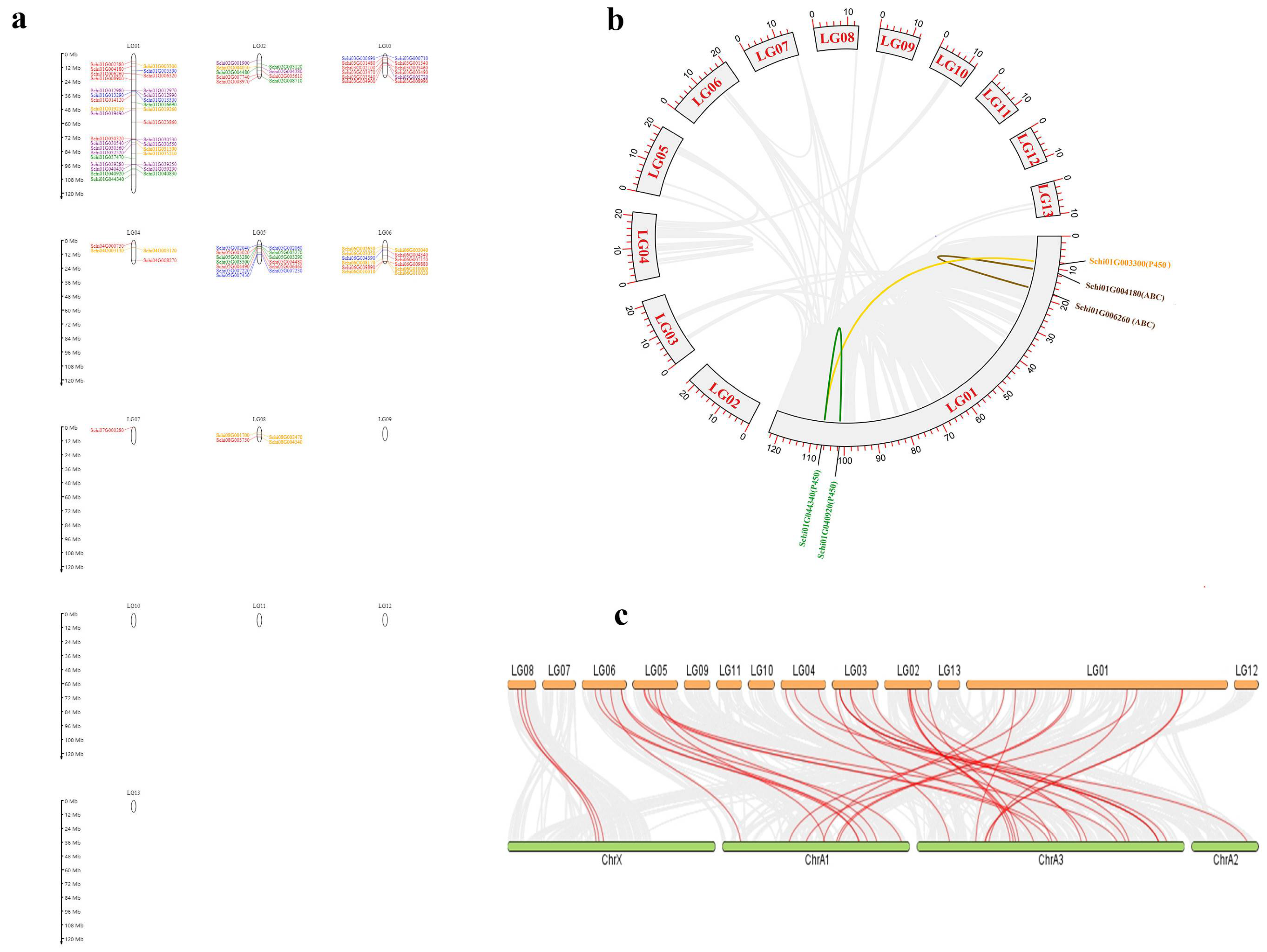
3.3. Chromosomal Location and Collinearity of Detoxification Genes of S. chinensis
The location and collinearity analysis of all detoxification genes showed that 144 genes were unevenly distributed on chromosomes 1–9 (Figure 7a). Chromosome 1 had the most members of detoxification genes with 33 genes, among which there were eight genes in ABCs, four genes in CCEs, three genes in GSTs, six genes in P450s and 12 genes in UDPs. A total of two pair genes showed tandem duplications on Chromosome 1. The two genes Schi01G040920 and Schi01G044340 of the P450 family, and Schi01G004180 and Schi01G00620 of the ABCs showed tandem duplications, respectively. Only two pairs of genes in the detoxification genes had collinearity (Figure 7b). Chromosome 7 had the fewest detoxification genes with only one member. The distribution of detoxification genes on chromosomes with no bias to the 5′ or 3′ ends may be related to their function.
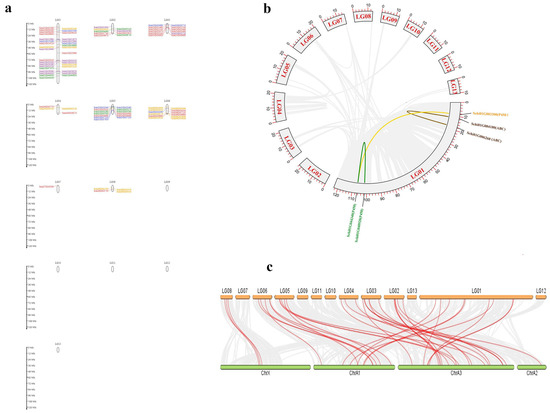
Figure 7.
Location and collinearity analysis of all detoxification genes in S. chinensis. (a) Scaffold location and gene tandem. Green represent CCEs; Blue represent GSTs; orange represent P450s; purple represent UDPs; Red represent ABCs (b) Chromosomal location and collinearity. Grey boxes represent chromosomes. lighted lines connected detoxification gene duplication. (c) Synteny on gene families of S. chinensis and A. pisum.
The collinear comparison map of the detoxification gene family was established by MC Scan X between S. chinensis and A. pisum (Figure 7c). There were 38 pairs of collinearity (homologous gene pairs) in A. pisum, including nine in P450s, 16 in ABCs, three in CCEs, three in GSTs, and seven in UDPs. There were more homologous gene pairs of ABCs and P450s in the S. chinensis and A. pisum, which may be related to the large number of these two gene families. It is inferred that these two gene families are relatively conservative in the evolutionary process and have relatively stable functions.
The Ka/Ks analysis of 38 pairs of homologous genes existing in S. chinensis and A. pisum was carried out, and the results are shown in Table 2. The ratios of Ka/Ks between gene pairs were all <1, which indicated that the detoxification genes in S. chinensis were mainly affected by purification selection during evolution.
Table 2.
Nucleotide substitution rate of detoxification genes in S. chinensis.
3.4. Expression Profiles of Detoxification Genes in S. chinensis
We examined 61 detoxification genes in the transcriptome data of S. chinensis, among which there were 13 genes in ABCs, nine genes in CCEs, seven genes in GSTs, 19 genes in P450s and 11 genes in UDPs, respectively. The gene number expressed in the P450s was the highest and the gene number expressed in GSTs was the least (Table 3). In terms of gene expression, the overall expression of CCEs was the highest, while that of P450s was the lowest. Individual gene expression was particularly high in some gene families. For example, the expression level of Schi01G005590 (GSTs) was 251.68, Schi01G003300 (P450s) was 166.92., and Schi05G003270(CCEs) was 127.19, from which we infer that these genes play an important role in the detoxification process of S. chinensis.
Table 3.
Expression profiles of detoxification genes in S. chinensis.
3.5. Physicochemical Properties of Detoxification Gene Products in S. chinensis
The molecular weight of the detoxification genes products ranged from 20,632.58 Da to 199,951.12 Da (Table 4), the number of amino acids from 178 to 1764, and the aliphatic index from 74.78 to 112.86. The isoelectric points (pI) ranged from 4.93 to 9.58. The instability index of which 51% of the detoxification gene products were more than 40, indicated that those genes were unstable and easily degraded. The aliphatic amino acid index of detoxification gene proteins was 74.78–112.86, and the average of the hydropathicity indices ranged from −0.81 to 0.306. The proteins with hydropathicity value less than 0 were hydrophilic proteins.
Table 4.
General information and physicochemical properties of detoxification gene family products in S. chinensis.
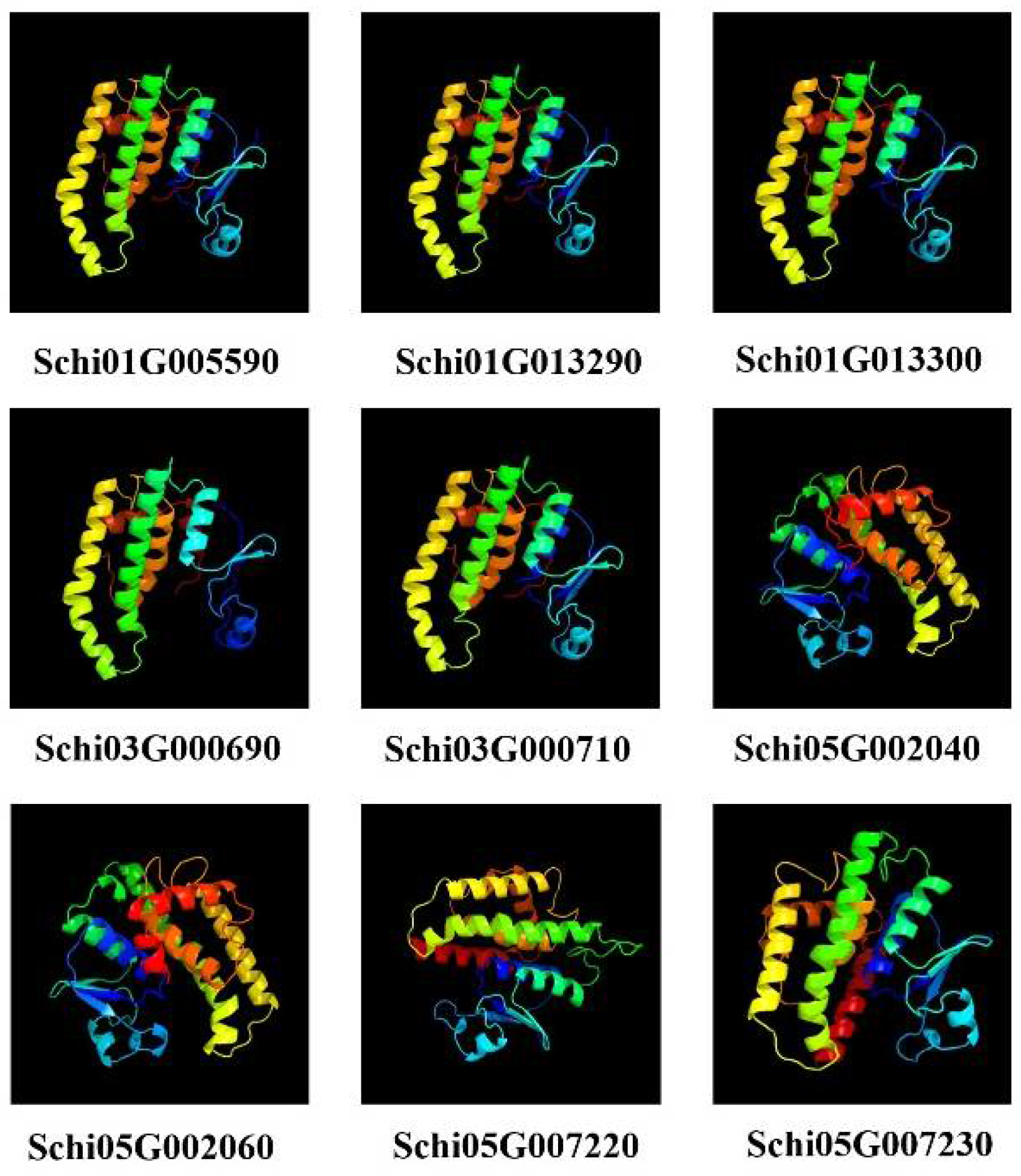
3.6. Prediction of Protein Multi-Level Structures of Detoxification Gene Products in S. chinensis
Since the members of detoxification gene family products have similar protein structures, we selected the protein members of GSTs to predict secondary and tertiary structures. The predicted secondary structures of the nine GST proteins are shown in Table 5. The GST protein was composed of four parts: α-helix, extended chain, β-turn and random coil, among which the α helix ratio of the GST protein was the highest, followed by random coil, and the ratio of β-turn was the smallest. Our Signal P prediction showed that there were no signal peptides in all the GST proteins of S. chinensis. The tertiary structures of GST proteins are shown in Figure 8. Schi01G005590, Schi01G013290, Schi01G013300, Schi03G000690 and Schi03G000710, Schi05G002040 and Schi05G002060, Schi05G007220, and Schi05G007230 have similar protein structures, respectively. These genes belonged to sigma, theta, and delta subfamilies, respectively. The result indicated that gene members belonging to the same subfamily have similar protein structures, indicating they have similar biological functions.
Table 5.
Secondary structure of the GST gene family proteins in S. chinensis.
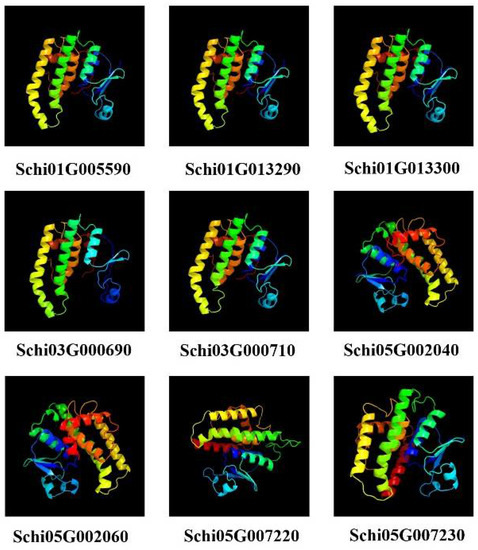
Figure 8.
Prediction of tertiary structures of GSTs proteins in S. chinensis.
4. Discussion
4.1. Expansion and Contraction of Detoxification Genes in S. chinensis
The sequenced genomes of A. pisum, M. persicae, and C. cedri with manually annotated detoxification gene families served as comparisons for our studies [50,51]. The number of detoxification genes in C. cedri was derived from the annotation table in NCBI with Accession No. GCA_902439185.1, and those of A. pisum and M. persicae were derived from published articles [16,52,53,54].
The number of detoxification genes of S. chinensis was 144, less than that of A. pisum, C. cedri and M. persicae, which might be related to its unique host plant. It was predicted that polyphagous insects require a greater complement of detoxification-related enzymes for they were usually exposed to a higher diversity of plant secondary metabolites than oligophagous ones [54,55]. The gene number of GSTs and UDPs in S. chinensis was nine and 14, respectively, which were relatively less and conserved by comparison with the other three aphids. The fewer detoxification-related genes may be due to the unobvious duplication events of detoxification-related genes. Genes with conserved roles usually have relatively stable copies, while those with diversified functions have higher rates of gain-and-loss with random changing degrees [56]. The numbers of ABCs of S. chinensis was 55, that was more than other aphids and showed significant genetic expansion. It is inferred that the ABCs play an important role in the degradation of secondary metabolites of S. chinensis. The number of P450s was 48 in S. chinensis, which was clearly contracted by comparison with the other three aphids. CYPs in many insects are associated with the metabolism or detoxification of key endogenous substrates and xenobiotics, such as steroid hormones and lipids, plant natural products, and pesticides, which are key components for the successful adaptation of insects to their host plants [57].
4.2. Characteristics and Expression of the Detoxification Genes of S. chinensis
The current study compared characteristics of the five detoxification gene families of S. chinensis with other three aphids, A. pisum, M. persicae, and C. cedri. Ten conserved motifs were found in four gene families, except for GSTs with six conserved motifs. The conserved motifs in detoxification genes are very important in the functional domains, and the highest one was its key structure, while the motif patterns can finely tune the function of detoxification genes [58]. Structural variation affects gene evolution [59]. The detoxification gene proteins encoded by the subfamily members usually had the same motif orders and exons, which indicated that genes in closely related groups were highly conserved and might have similar functions.
4.3. Collinearity, Chromosome Position and Evolutionary Rate of Detoxification Genes of S. chinensis
We performed analysis on collinearity relationships to further investigate the gene duplication events within detoxification genes. There were two pairs of gene duplications in the detoxification genes of S. chinensis, including P450s and ABCs. The differential expansion events during the species evolution might result in the phenomena that the number of family members was not correlated with genome size. Tandem duplications generate a large number of genes, which is considered as the most effective mechanism for producing and maintaining gene copies [57]. It was reported that gene duplications were critical for the evolution of new genes and novel functions and were major forces driving gene family expansion [60]. For example, CYP genes, often clustering in genomes, were considered as a result of gene duplication events [56]. We calculated the Ka and Ks values to estimate the evolutionary trend and revealed a functional selection pressure between duplicated gene pairs. The ratio of Ka to Ks in protein-coding genes can determine whether there is selection pressure acting in the process of gene evolution. Detoxification genes of S. chinensis showed a strong purifying selection during evolution, which suggested that their functions may be evolutionarily conserved.
5. Conclusions
Here, we performed a comprehensive genome-wide analysis of the detoxification gene family in S. chinensis and compared the results with the genomes of A. pisum, M. persicae, and C.cedri. We manually annotated 144 genes of S. chinensis, including nine in GSTs, 55 in ABCs, 18 in CCEs, 48 in P450s, and 14 in UDPs. We constructed the phylogenetic trees, motif patterns, domains, and gene structures of detoxification genes from these four aphids and further analyzed the chromosomal location, collinearity, evolution rates, and their expression. Finally, we predicted characteristics, physicochemical properties, and protein multi-level structures of detoxification gene products of S. chinensis. Our results provide comprehensive information, molecular data, and gene candidates for further analyses. S. chinensis can survive in galls with the tannin content up to 70%, and we infer that it has a strong ability to reduce secondary metabolites. The phenomenon may be related not only to its own detoxification genes, but also to the existence of endosymbionts or a long-term obligate parasitic relationship.
Author Contributions
H.H. and Z.R. designed the study, interpreted all the data and findings and wrote the manuscript, and made equal contributions as major authors, while M.J.C.C. validated, revised, and edited the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was partially supported by The National Natural Science Foundation of China (31870366), Shanxi International Science and Technology Cooperation Project (201803D421051), Research Project Supported by Shanxi Scholarship Council of China (2020-018), the National High Technology Research and Development “863” Program (2014AA021802).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The protein coding sequences of detoxification genes of the three aphid species, Acyrthosiphon pisum, Cinara cedri and Myzus persicae, were downloaded from the Insect BASE website (http://v2.insect-genome.com/, accessed on 12 May 2022).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bell, J. Chinese galls. Pharmaceut. J. 1851, 10, 128. [Google Scholar]
- Tang, C.; Tsai, P.H. Studies on the Chinese gallnuts of meitan, Kweichow. Acta Entomol. Sin. 1957, 7, 131–142. [Google Scholar]
- Zhang, G.X.; Qiao, G.X.; Zhong, T.S.; Zhang, W.Y. Fauna Sinica Insecta. Homoptera: Mindaridae and Pemphigidae; Science Press: Beijing, China, 1999; p. 14. [Google Scholar]
- Baker, A.C. On the Chinese gall (Aphididae-Hom). Ent. News. 1917, 28, 385–393. [Google Scholar]
- Li, Z.G.; Yang, W.Y.; Xia, D.J. Study on the Chinese gallnuts. For. Res. 2003, 16, 760–767. [Google Scholar]
- Blackman, R.L.; Eastop, V.F. Aphids on the World’s Crops: An Identification and Information Guide; John Wiley and Sons: New York, NY, USA, 1984. [Google Scholar]
- Heie, O.E. The Aphidoidea (Hemiptera) of Fennoscandia and Denmark. I. General part, the families Mindaridae, Hormaphididae, Thelaxidae, Anoeciidae, and Pemphigidae. Fauna Entomol. Scand. 1980, 9, 206–207. [Google Scholar]
- Remaudière, G.; Remaudière, M. Catalogue of the World’s Aphididae (Homoptera Aphidoidea); INRA: Paris, France, 1997. [Google Scholar]
- Zhang, G.X.; Zhong, T.S. Economic Insect Fauna of China, Fasc. 25, Homoptera: Aphidinea; Science Press: Beijing, China, 1983. (In Chinese) [Google Scholar]
- Yang, Z.X.; Chen, X.M.; Nathan, H.; Feng, Y. Phylogeny of Rhus gall aphids (Hemiptera: Pemphigidae) based on combined molecular analysis of nuclear EF1a and mitochondrial COII genes. Entomol. Sci. 2010, 13, 351–357. [Google Scholar] [CrossRef]
- Chen, M.S. Inducible direct plant defense against insect herbivores: A review. Insect Sci. 2008, 15, 101–114. [Google Scholar] [CrossRef]
- Dermauw, W.; Van-Leeuwen, T. The ABC gene family in arthropods: Comparative genomics and role in insecticide transport and resistance. Insect Biochem. Mol. Biol. 2014, 45, 89–110. [Google Scholar] [CrossRef]
- Després, L.; David, J.P.; Gallet, C. The evolutionary ecology of insect resistance to plant chemicals. Trends Ecol. Evol. 2007, 22, 298–307. [Google Scholar] [CrossRef]
- Li, X.; Schuler, M.A.; Berenbaum, M.R. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu. Rev. Entomol. 2007, 52, 231–253. [Google Scholar] [CrossRef]
- Urlacher, V.B.; Girhard, M. Cytochrome P450 Monooxygenases in Biotechnology and Synthetic Biology. Trends Biotechnol. 2019, 37, 882–897. [Google Scholar] [CrossRef] [PubMed]
- Chertemps, T.; Le-Goff, G.; Maïbèche, M.; Hilliou, F. Detoxification gene families in Phylloxera: Endogenous functions and roles in response to the environment. Comp. Biochem. Physiol. Part D Genom. Proteom. 2021, 40, 100867. [Google Scholar] [CrossRef] [PubMed]
- Oakeshott, J.G.; Devonshire, A.L.; Claudianos, C.; Sutherland, T.D.; Horne, I.; Campbell, P.M.; Ollis, D.L.; Russell, R.J. Comparing the organophosphorus and carbamate insecticide resistance mutations in cholin- and carboxyl-esterases. Chem. Biol. Interact. 2005, 157, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.M.; Xu, B.Y.; Si, F.L.; Li, J.; Yan, Z.T.; Yan, Z.W.; He, X.; Chen, B. Identification of carboxylesterase genes associated with pyrethroid resistance in the malaria vector Anopheles sinensis (Diptera: Culicidae). Pest Manag. Sci. 2018, 74, 159–169. [Google Scholar] [CrossRef]
- Jakoby, W.B.; Ziegler, D.M. The enzymes of detoxication. J. Biol. Chem. 1990, 265, 20715–20718. [Google Scholar] [CrossRef]
- Shi, H.; Pei, L.; Gu, S.; Zhu, S.; Wang, Y.; Zhang, Y.; Li, B. Glutathione S-transferase (GST) genes in the red flour beetle, Tribolium castaneum and comparative analysis with five additional insects. Genomics 2012, 100, 327–335. [Google Scholar] [CrossRef]
- Cheng, T.; Wu, J.; Wu, Y.; Chilukuri, R.V.; Huang, L.; Yamamoto, K. Genomic adaptation to polyphagy and insecticides in a major East Asian noctuid pest. Nat. Ecol. Evol. 2017, 1, 1747–1756. [Google Scholar] [CrossRef]
- Xiao, L.F.; Zhang, W.; Jing, T.X.; Zhang, M.Y.; Miao, Z.Q.; Wei, D.D.; Yuan, G.R.; Wang, J.J. Genome-wide identification, phylogenetic analysis, and expression profiles of ATP-binding cassette transporter genes in the oriental fruit fly, Bactrocera dorsalis (Hendel) (Diptera: Tephritidae). Comp. Biochem. Physiol. Part D Genom. Proteom. 2018, 25, 1–8. [Google Scholar] [CrossRef]
- Geer, L.Y.; Geer, R.C.; Gonzales, N.R. CDD: A conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, 225–229. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bolger, A.M.; Marc, L.; Bjoern, U. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 15, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.Y.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene features visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 289660. [Google Scholar] [CrossRef]
- Wang, Y.P.; Tang, H.B.; Debarry, J.D.; Tan, X.; Li, J.P.; Wang, X.Y.; Lee, T.; Jin, H.Z.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.Q.; Wang, J.; Wong, G.K.; Yu, J. Ka Ks_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinf. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Wang, D. An Improved TRIzol Method to Extract Total RNA from Skin Tissue of Rana dybowskii. Chin. J. Wildl. 2012, 33, 127–128. [Google Scholar]
- Sharon, D.; Tilgner, H.; Grubert, F.; Snyder, M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [Google Scholar] [CrossRef]
- Mak, S.; Gopalakrishnan, S.; Car, E.C. Comparative performance of the BGISEQ-500 vs Illumina HiSeq2500 sequencing platforms for palaeogenomic sequencing. Giga Sci. 2017, 6, gix049. [Google Scholar] [CrossRef] [PubMed]
- Mckinney, G.J.; Hale, M.C.; Goetz, G.; Gribskov, M.; Thrower, F.P.; Nichols, K.M. Filtered trinity assembly. 2015, 8, 1494–1512. [Google Scholar]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P. Integrated NR Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 132, D115–D119. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, gkt1223. [Google Scholar]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Kazutaka, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 13, 1933–1942. [Google Scholar]
- Ison, J.; Kalas, M.; Jonassen, I.; Bolser, D.; Uludag, M.; McWilliam, H.; Malone, J.; Lopez, R.; Pettifer, S.; Rice, P. EDAM: An ontology of bioinformatics operations, types of data and identifiers, topics and formats. Bioinformatics 2013, 15, 1325–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almagro-Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Geourjon, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 1995, 11, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.S.; Rider, D.S.; Walsh, T.K.; de Vos, M.; Gordon, K.H.; Ponnala, L.; Macmil, S.L.; Roe, B.A.; Jander, G. Comparative analysis of detoxification enzymes in Acyrthosiphon pisum and Myzus persicae. Insect Mol. Biol. 2010, 9, 155–164. [Google Scholar] [CrossRef]
- Julca, I.; Marcet-Houben, M.; Cruz, F.; Vargas-Chavez, C.; Johnston, J.S.; Gómez-Garrido, J.; Frias, L.; Corvelo, A.; Loska, D.; Cámara, F.; et al. Phylogenomics Identifies an Ancestral Burst of Gene Duplications Predating the Diversification of Aphidomorpha. Mol. Biol. Evol. 2020, 37, 730–756. [Google Scholar] [CrossRef]
- Feyereisen, R. Arthropod CYP omes illustrate the tempo and mode in P450 evolution. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 19–28. [Google Scholar] [CrossRef]
- Dermauw, W.; van-Leeuwen, T.; Feyereisen, R. Diversity and evolution of the P450 family in arthropods. Insect Biochem. Mol. Biol. 2020, 127, 103490. [Google Scholar] [CrossRef]
- Mathers, T.C.; Chen, Y.; Kaithakottil, G.; Legeai, F.; Mugford, S.T.; Baa-Puyoulet, P. Rapid transcriptional plasticity of duplicated gene clusters enables a clonally reproducing aphid to colonize diverse plant species. Genome Biol. 2017, 18, 27. [Google Scholar] [CrossRef]
- Yates, A.D.; Michel, A. Mechanisms of aphid adaptation to host plant resistance. Curr. Opin. Insect Sci. 2018, 26, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Yang, M.; Yao, B. The phylogenetic and evolutionary analyses of detoxification gene families in Aphidinae species. PLoS ONE 2022, 17, e0263462. [Google Scholar] [CrossRef] [PubMed]
- Heidel-Fischer, H.M.; Vogel, H. Molecular mechanisms of insect adaptation to plant secondary compounds. Curr. Opin. Insect Sci. 2015, 8, 8–14. [Google Scholar] [CrossRef]
- Xu, Y.L.; He, P.; Zhang, L.; Fang, S.Q.; Dong, S.L.; Zhang, Y.J.; Li, F. Large-scale identification of odorant-binding proteins and chemosensory proteins from expressed sequence tags in insects. BMC Genom. 2009, 10, 632. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Shi, F. Evolution of the RALF gene family in plants: Gene duplication and selection patterns, Evol. Bioinf. 2012, 8, 271–292. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).