Abstract
The restricted rotation of chemical bonds may lead to the formation of stable, conformationally chiral molecules. While the asymmetry in chiral molecules is generally observed in the presence of one or more stereocenters, asymmetry exhibited by conformational chirality in compounds lacking stereocenters, called atropisomerism, depends on structural and temperature factors that are still not fully understood. This atropisomerism is observed in natural diarylether heptanoids where the length of the intramolecular tether constrains the compounds to isolable enantiomers at room temperature. In this work, we examine the impact tether length has on the activation free energies to isomerization of a diarylether cyclophane substructure with a tether ranging from 6 to 14 carbons. Racemization activation energies are observed to decay from 48 kcal/mol for a 7-carbon tether to 9.2 kcal/mol for a 14-carbon tether. Synthetic efforts to experimentally test these constraints are also presented. This work will likely guide the design and synthesis of novel asymmetric cyclophanes that will be of interest in the catalysis community given the importance of atropisomeric ligands in the field of asymmetric catalysis.
1. Introduction
The asymmetry present in chiral molecules is central to innovations in chemistry, molecular biology, and pharmaceutical development. The fact that enantiomers of chiral drugs may have differing activities ranging from being ineffective to biologically harmful particularly emphasizes the importance of chiral synthesis or separation of chiral intermediates within the medicinal drug pipeline [1]. Although molecular chirality is commonly discussed in terms of stereogenic elements, the restricted rotation around a single bond may also render a molecule kinetically inconvertible between two separable, asymmetric mirror image conformations (termed atropisomers) [2,3,4]. There are a vast number of natural and synthetic compounds that are conformationally chiral [3,5,6,7]. BINOL and BINAP are prototypical examples of these kinds of molecules.
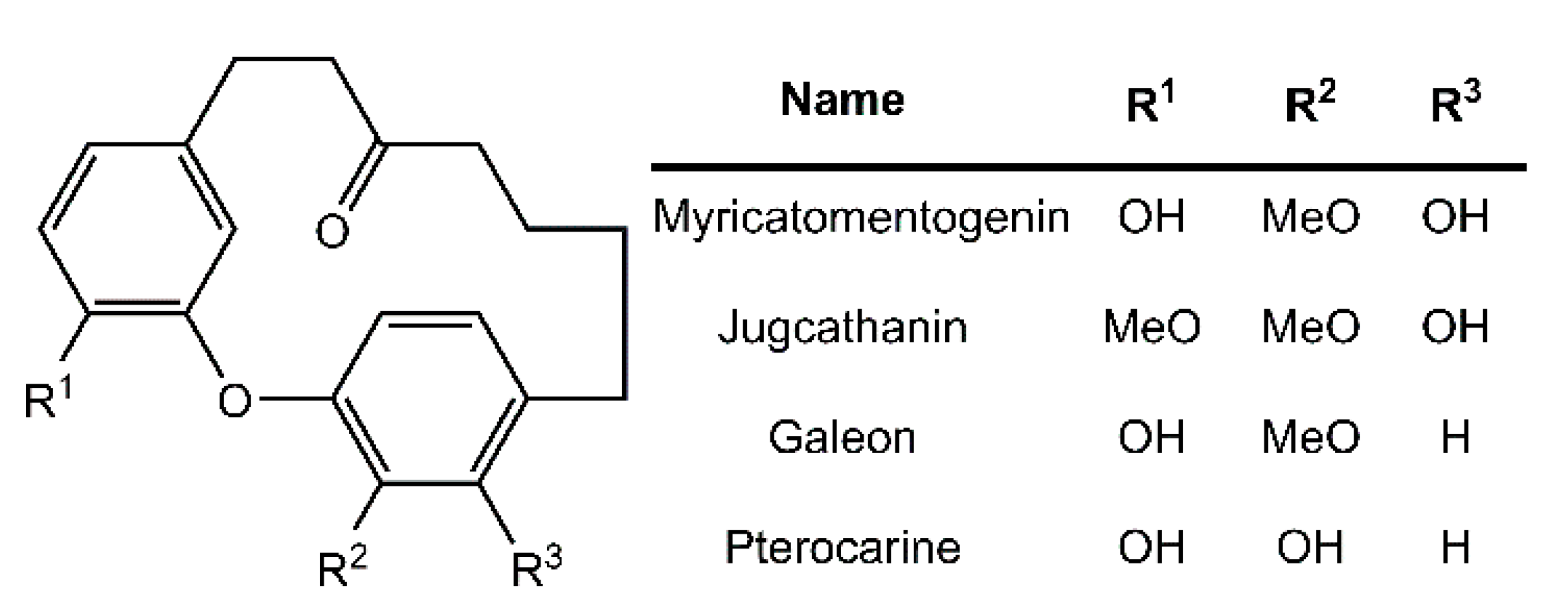
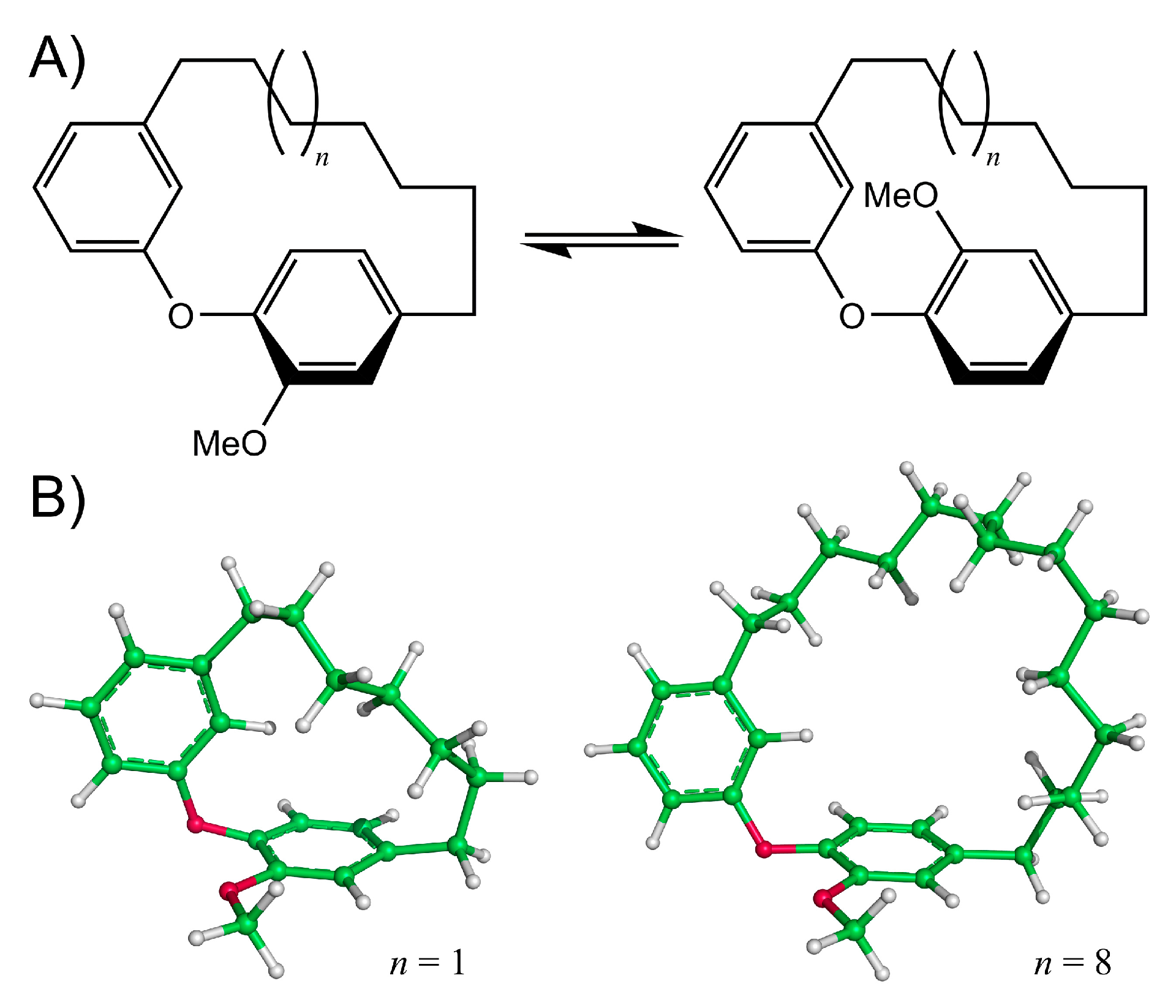
Diarylether heptanoids such as myricatomentogenin, jugcathanin, galeon, and pterocarine (Figure 1) are examples of chiral, naturally occurring, asymmetric plant metabolites with biological properties of medicinal interest (hepatoprotective activity, anti-androgenic activity, inhibition of nitric oxide production, anti-inflammatory activity, and anti-tumor activity) [8,9,10]. The atropisomerism of these compounds arises from the hindered rotation of the para-substituted diarylether ring sterically clashing with an intramolecular tether (or “rope”). The seven-carbon tether is short enough to restrict the “rope-skipping” isomerization (Figure 2A) of the molecules, but longer lengths may be expected to permit racemization (Figure 2B) [11]. Collectively, the unique structure and biological activities of these diarylethyl heptanoids open questions into the length of the tether required to maintain isolatable enantiomers of tethered diarylethers along with the effect the tether may have on molecular binding to biological targets. To date, no systematic investigation of the relationship between tether length and barrier to racemization in diarylether cyclophanes has been reported.
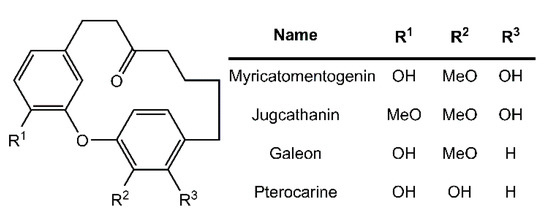
Figure 1.
Structure of select diarylethyl heptanoids.
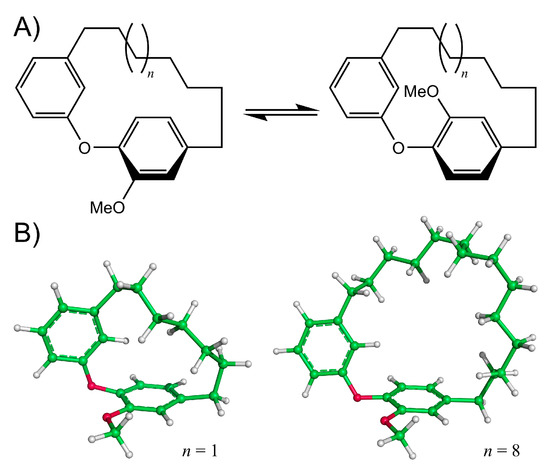
Figure 2.
(A) Rope-skipping isomerization of a diarylether cyclophane structure. (B) Structures of the diarylether cyclophane with a 7-carbon tether (left, n = 1) and a 14-carbon tether (right, n = 8).
Preliminary insight into the relationship between tether length and isomerization may be found in studies investigating the racemization of simpler cyclophane structures containing a single aromatic ring rather than a diarylether core [12]. Work by Koenig and coworkers on paracyclophanes reported a 10-carbon tether was short enough to prevent isomerization, while a 12-carbon tether readily racemized above 40 °C [11]. Research by Kanomata has similarly reported the synthesis of enantiomeric cyclophanes with 10 and 11 carbons isolable at room temperature [13]. Quantitatively similar results have also been found with 1,n-dioxa[n]paracyclophanes and 1,n-diaza[n]paracyclophanes [14,15]. Whether similar tether lengths will yield comparably isolable atropisomers at room temperature for diarylethyl cyclophanes remains unknown and is the point of this investigation. The knowledge gained from understanding the relationship between intramolecular tether and diarylether isomerization may help guide synthesis of chiral, conformationally constricted drugs or ligands with unprecedented chiral diarylether topologies. Efforts toward the syntheses of diarylether cyclophanes of varying tether lengths will also be discussed to guide future work and to encourage other labs to pursue this chemistry further.
2. Methods
Models of myricatomentogenin, jugcathanin, galeon, and pterocarine (Figure 1) were constructed along with the diarylether cyclophane (DAEC) skeleton structure with aliphatic chain lengths ranging from 6 (Figure 2, n = 0) to 14 (n = 8) CH2 units (Figure 2). In consideration of the flexibility of the aliphatic chain, varying conformations of the molecules were sampled using the random incremental pulse search (RIPS) stochastic search algorithm using the Molecular Operating Environment (MOE) software package [16,17]. The RIPS algorithm operates by perturbing rotatable bonds and then performing a geometry relaxation. Default parameters for this stochastic search in MOE were employed where the energy minimization used the MMFF94x force field and incorporated a distance-dependent implicit solvation effect (dielectric constant ε = 80).
A portion of the RIPS conformers representing a major fraction of the Boltzmann population (Table 1) were further geometrically optimized at the quantum mechanical (QM) level of theory using density functional theory with the hybrid B3LYP exchange-correlation functional and 6-31G(d’) basis set [18,19,20]. These computations included implicit solvation via the solvation model based on density (SMD) method using the dielectric constant of benzene (ε = 2.2706) [21]. For each of the molecules investigated, the 7 lowest-energy, unique QM-optimized conformations were selected as starting geometries for the isomerization transition state (TS) search; the most stable conformation identified by the RIPS algorithm was also included as a starting geometry if it was not one of the 7 initially selected structures. Recomputing the mole fraction for these 7 structures among the QM-optimized, unique structures indicates that these 7 conformers account for the majority of the ensemble conformations for jugcathanin, pterocarine, myricatomentogenin, galleon, and DAECn=1,2 (Table 1). Increasing the DAEC tether increases molecular flexibility and accessible conformations, and it is consequently observed that using the 7 most stable conformers accounts for a reduced but reasonable portion of the ensemble. It is not within the scope of this work to seek Boltzmann averaged results for these molecules, though we do not anticipate substantial differences between such values and what is reported here.

Table 1.
Mole fractions for selected molecular conformers at 298 K.
Transition state searches with an SMD medium simulating both benzene and dimethylformamide (DMF, ε = 37.219), a likely candidate solvent for experimental synthesis of these molecules, were conducted. Instances where TS searches from different starting geometries converged to identical maxima and instances where TS searches were not able to successfully isolate the appropriate TS structure led to size reduction of the presented data. All quantum mechanical computations were performed using Gaussian16 software [22].
3. Results and Discussion
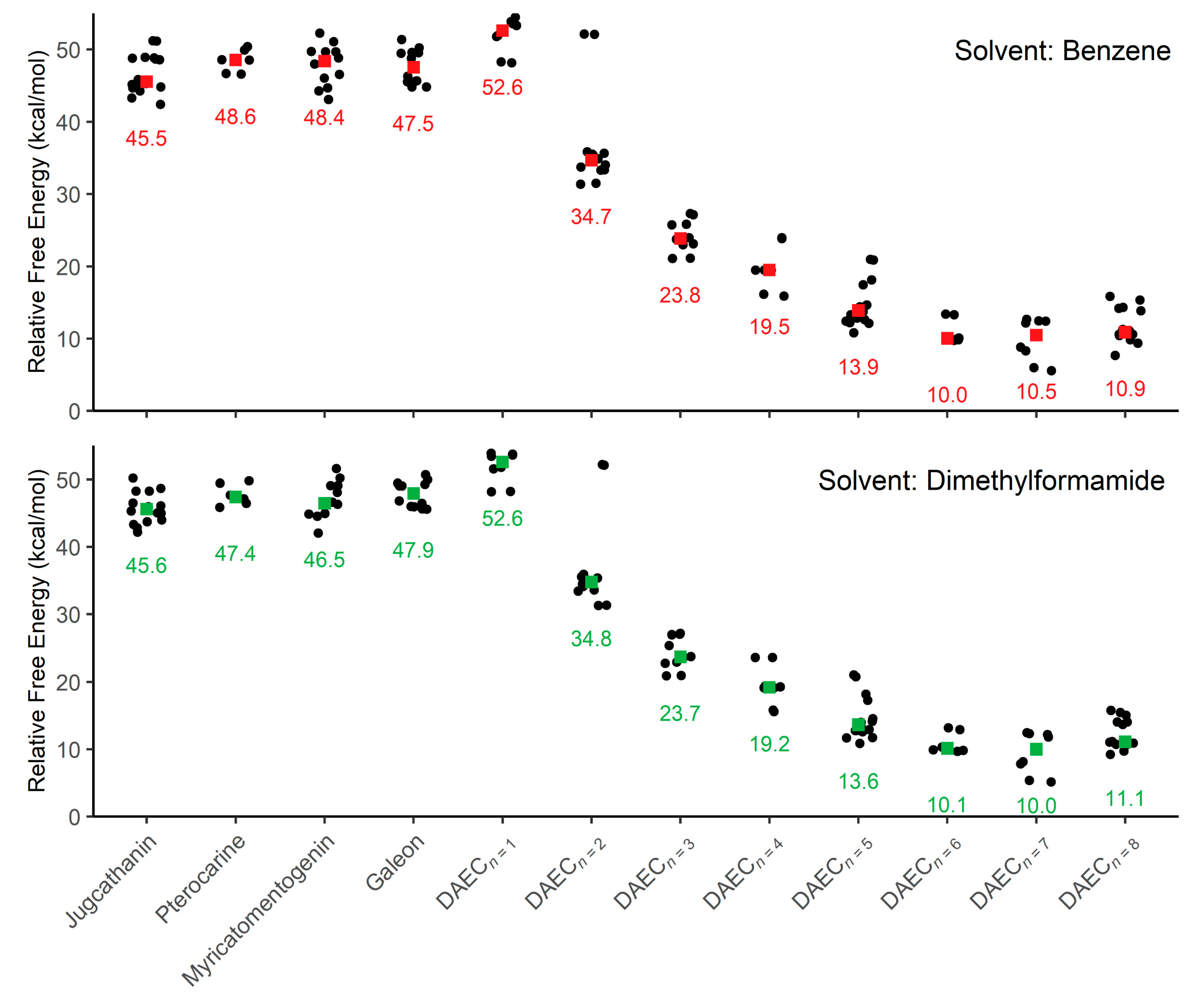
This computational study focuses on elucidating the energetic details of the isomerization of four diarylether heptanoid structures and a diarylether cyclophane structure with varying tether lengths (Figure 2A, 6 + n, where n = 0 to 8). Plots of the relative activation free energies (ΔG‡) computed for the isomerization of both enantiomer conformations (i.e., both forward and reverse isomerization reaction energies) for all compounds are illustrated in Figure 3.
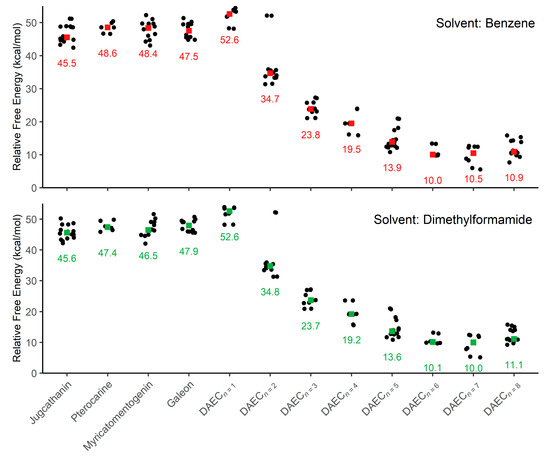
Figure 3.
Plots of relative activation energies for isomerization between enantiomeric conformations within an implicit solvent of benzene and dimethylformamide. Colored squares are used to indicate the median activation energy (also listed) for each sample of molecule conformations.
Focusing first on the four diarylether heptanoid compounds, the lowest isomerization activation free energies computed with an SMD medium of benzene are 42.4 kcal/mol for jugcathanin, 46.6 kcal/mol for pterocarine, 43.1 kcal/mol for myricamentogenin, and 44.8 kcal/mol for galeon. Use of DMF as the SMD medium is observed to generally reduce the required activation energy for the lowest energy TS pathway, with net shifts of ΔG‡ ≤ 1.1 kcal/mol for each of the molecules: 42.2 kcal/mol for jugathanin, 45.9 kcal/mol for pterocarine, 42.0 kcal/mol for myricamentogenin, and 45.6 kcal/mol for galeon. It is noted that these results are in striking agreement with work by Beaudry, Cheong, and coworkers who computed the ΔG‡ for racemization of galeon to be 44.5 kcal/mol (at the B3LYP/6-31G* level of theory with PCM implicit solvation using standard dichlorobenzene parameters) and predict the jugcathanin, pterocarine, and myricatomentogenin would have a similar ΔG‡ [23].
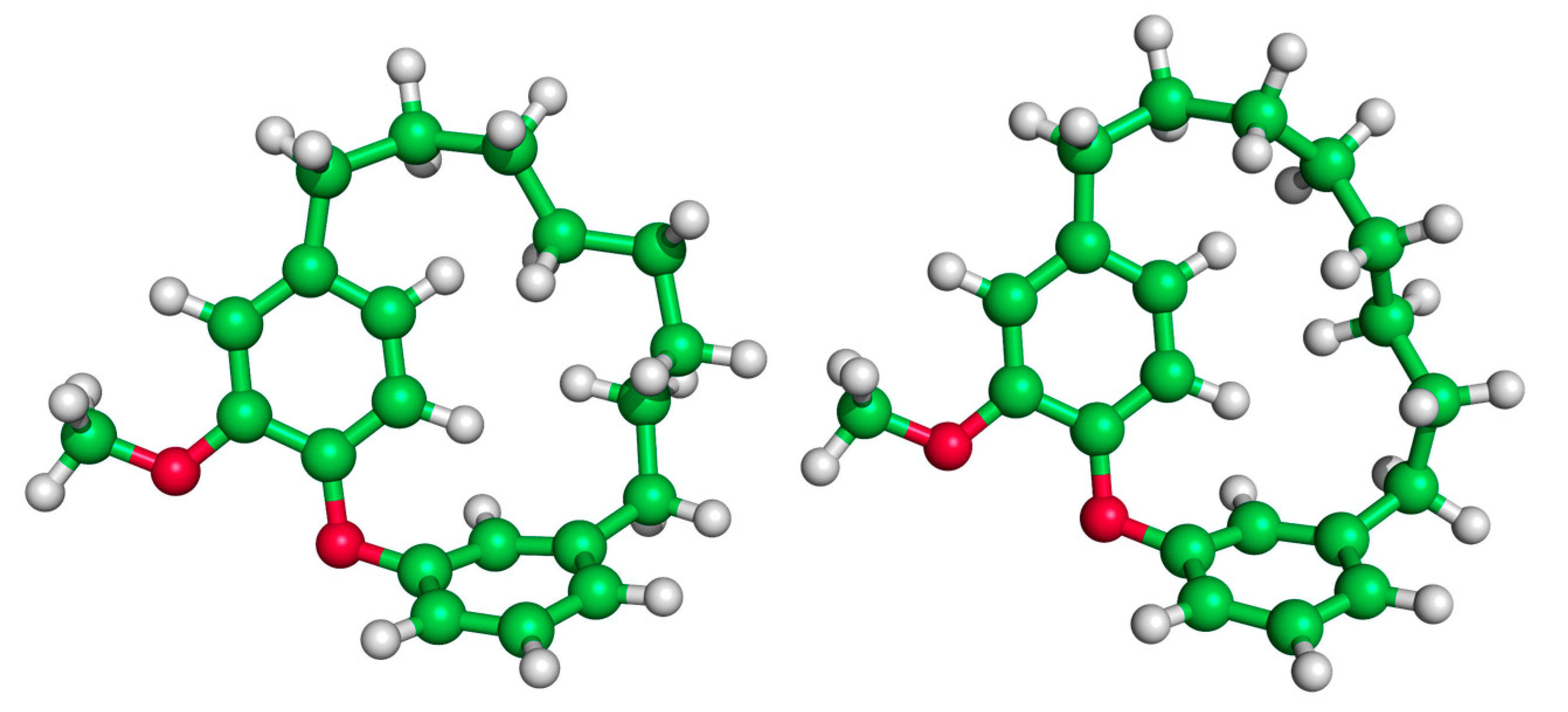
We now consider the DAEC structure with its tether length varying from 6 (Figure 2A, n = 0) to 14 (n = 8) carbons. Although energetic minima for conformations of the six-carbon DAECn=0 were successfully computed, no TS structures for the rope-skipping isomerization were able to be isolated as the tether is prohibitively short. Racemization TS structures were computed for DAECn=1 through DAECn=8 (Figure 3). In going from DAECn=1 to DAECn=2, the conformer with the lowest ΔG‡DMF is shown to significantly decrease from 48.1 kcal/mol to 31.3 kcal/mol. Our TS sampling isolated a high energy isomerization for DAECn=2 comparable in relative energy and structure to conformations regularly observed for DAECn=1. The increased energy barrier arises from that isomer TS structure having significant steric clashing between the rings and the particular tether conformation (Figure 4). Additional TS searches starting from higher energy conformers identified in the RIPS search would reveal a wider range of activation energy pathways, but we expect that the larger statistical ensemble of conformers would not significantly improve the results to be worth the significant increase in computational costs and labor.
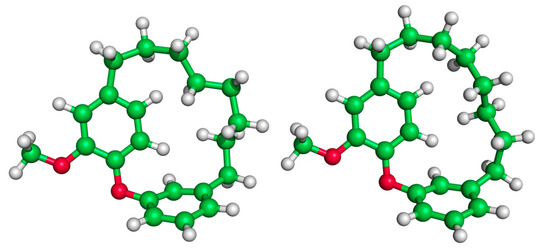
Figure 4.
Illustration of the high energy TS of DAECn=2 (left, ΔG‡ = 52.1 kcal/mol) compared to a low energy TS (right, ΔG‡ = 31.4 kcal/mol). The movement of the rings of the high energy TS structure is hindered by steric clashing between the ring and interlocking tether hydrogens.
As the tether is lengthened, the lowest ΔG‡ follows a power-law decay (Figure S1 in Supplementary Materials) where the isomerization barrier rapidly decreases until DAECn=5, beyond which the activation energies are generally consistent (ΔG‡DMF = 10.9 kcal/mol, 9.7 kcal/mol, 5.2 kcal/mol, and 9.2 kcal/mol for n = 5 through 8, respectively). This trend is independent of the implicit solvent used, as seen by the difference between median ΔG‡Benzene and ΔG‡DMF being ≤0.5 kcal/mol for each molecule (Figure 3). The lowest ΔG‡ for DAECn=7 is notably 4 kcal/mol lower than the other aforementioned structures; however, this difference arises because the two enantiomeric minima interlinked by this TS are 3.0 and 3.2 kcal/mol higher in free energy compared to the most stable conformer sampled. These results illustrate how isomerization change is also achievable by accessing higher energy conformations before the enantiomeric rope-skip occurs, though the effective activation energy for racemization remains similar. We have not explored interconversion pathways for conformational change prior to the rope-skip TS. Similar multistep pathways are expected for DAECn=8, though it was not observed in the TS searches sampled.
Two main synthetic strategies have been previously employed to prepare conformationally constrained diarylether cyclophanes. The most common strategy, especially in the preparation of the aforementioned natural products myricatomentogenin, jugcathanin, galleon, and pterocarine is to install the diarylether bond using an Ullmann coupling [9,24]. In contrast, a higher yielding SNAr approach was employed in the synthesis of the acerogenins [25]. Both approaches were pursued to ascertain whether either could lead to synthetically useful quantities of diarylether cyclophanes of varying tether lengths from which rotational barriers could be measured experimentally.
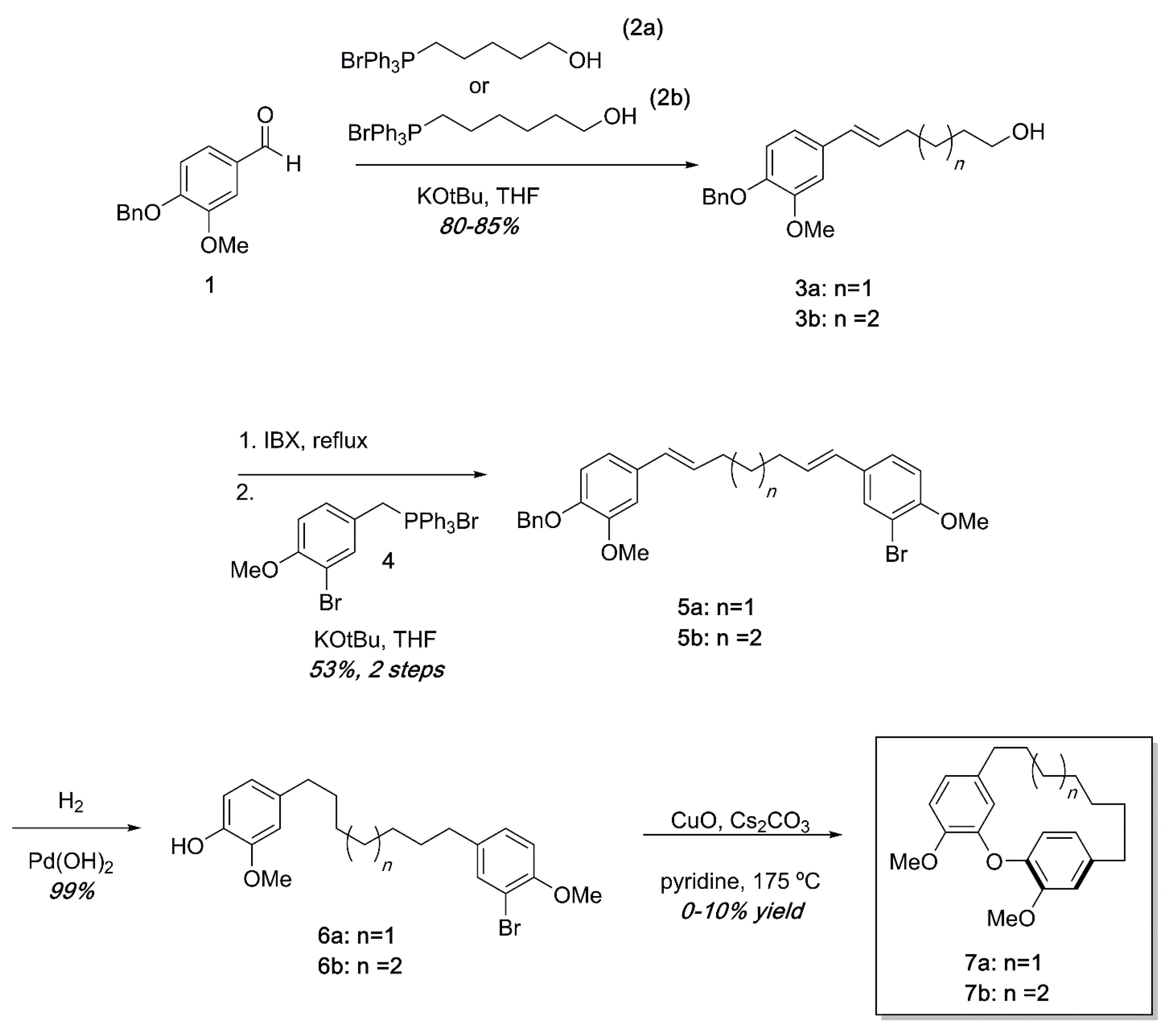
Ullmann substrates were prepared containing varying tether lengths using successive Wittig reactions as shown in Scheme 1. Beginning with commercially available 1 and the known Wittig reagents 2a and 2b [26], styrenes 3a and 3b could easily be accessed in high yields. Oxidation of the primary alcohol [27] and a second Wittig reaction with the known Wittig salt 4 led to dienes 5 in modest yields. Hydrogenation of 5a or 5b proceeded quantitatively to deliver the Ullmann substrates 6a and 6b. Ullmann coupling was attempted using various conditions including using K2CO3 or Cs2CO3 as base, temperatures from 130–210 °C, and Pd(OAc)2 catalysis [28]. After extensive reaction optimization, we found that the cyclization occurred in 0–10% yields in pyridine using CuO as a catalyst and Cs2CO3 as a base in a sealed tube at 175 °C. To avoid intermolecular Ullmann coupling, each reaction required high dilutions (0.005 M or less). In previous synthetic efforts, similar Ullman couplings with low yields are not unprecedented [24]. An additional frustration was that the high heat required for the reactions led to complex mixtures that were difficult to separate by chromatography. The combination of high dilution, low yields, and complex reaction mixtures made isolation of reasonable quantities of diarylether cyclophanes unsustainable by this synthetic pathway.
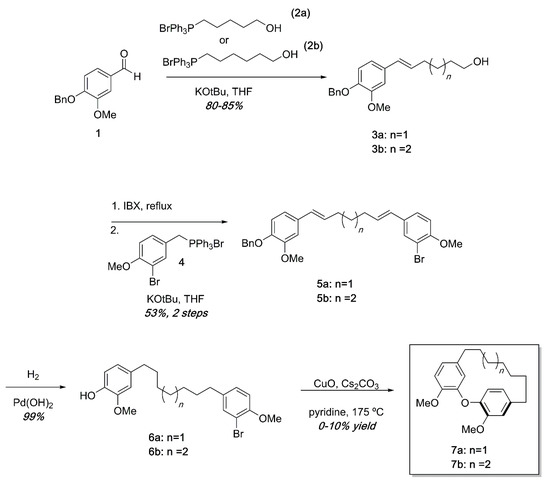
Scheme 1.
Synthesis of diarylether cyclophanes of varying tether lengths. While small amounts of cyclophanes were obtained, low yields, high dilution, and complex reaction mixtures in the final step made this approach impractical.
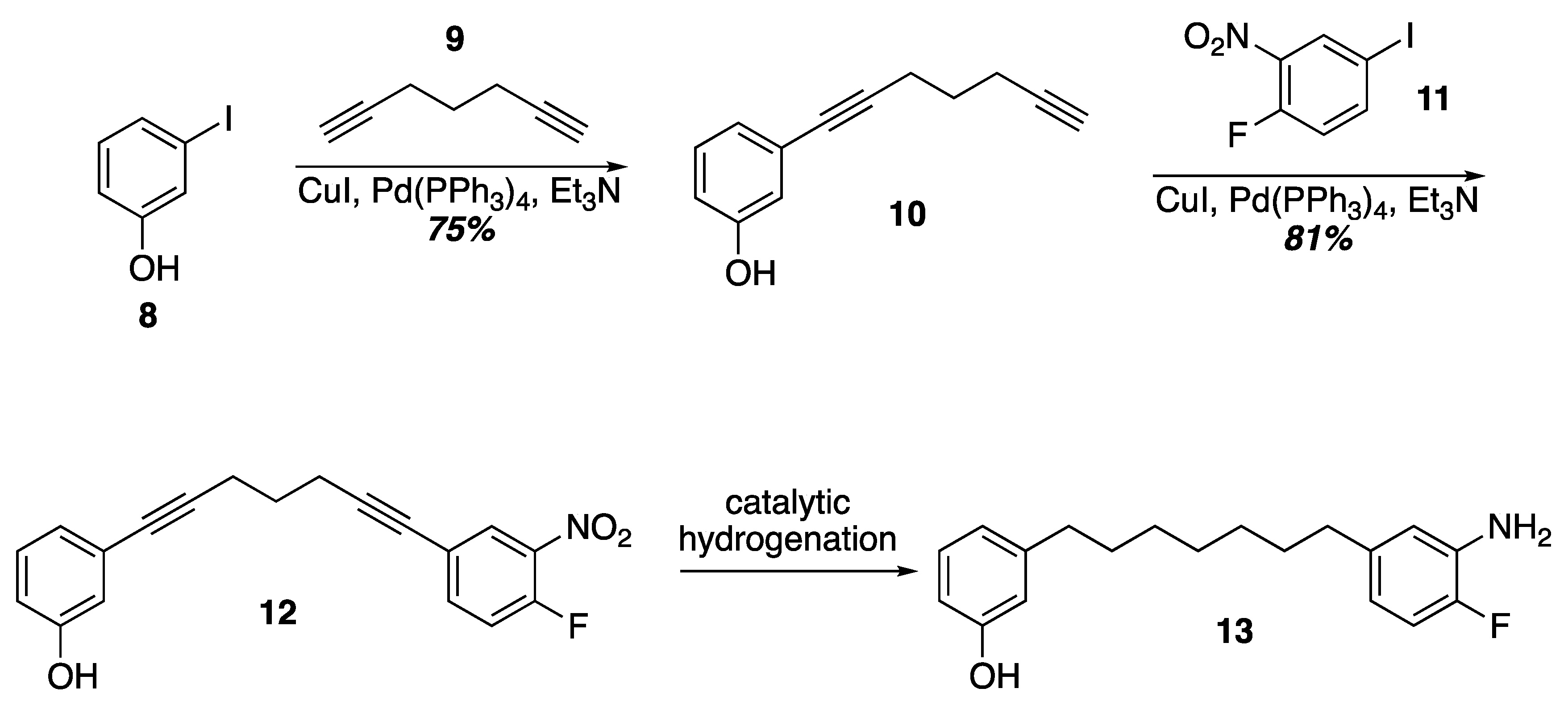
Prior work on the synthesis of the acerogenins showed that a final SNAr of a phenol on to an ortho-fluroronitro arene as the key cyclophane forming step could be executed at higher concentrations (0.1–1 M) and at room temperature. Given that terminal diynes from five to eleven carbons are commercially available as well as iodides 8 and 11, one idea to quickly construct SNAr substrates of various tether lengths would be to employ successive Sonogashira couplings followed by hydrogenation of the alkynes (Scheme 2). In fact, Sonogashira coupling of 3-iodophenol and 1,6-heptadiyne was uneventful leading to the terminal alkyne 10. Another Sonogashira coupling with commercially available 11 led to 12. Unfortunately, selective reduction of the alkynes in the presence of the nitro group with Wilkinson’s catalyst [29] led to indiscriminate reduction and attempts to re-oxidize the aniline back to a nitro group with MCPBA gave complex mixtures from which the target was not isolated [30]. Current efforts are underway to identify a viable route to these SNAr substrates that would allow varying tether lengths.
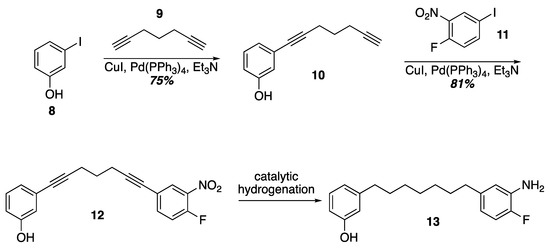
Scheme 2.
A double Sonogashira approach to an SNAr substrate.
4. Conclusions
Quantum mechanical computations were conducted to predict the activation energies for rope-skipping TSs for 4 diarylether heptenoid compounds and 8 diarylether cyclophane compounds with intramolecular tether chains ranging from 7 to 14 carbon atoms. The activation barriers for the diarylether heptenoids and DAECn=1 conformers sampled in DMF implicit solvation range from 42 to 54 kcal/mol and are in excellent agreement with previously reported values. Use of DMF as an alternative implicit solvation medium typically has a negligible (on average 0.7 kcal/mol) effect on predicted relative energies. Increasing the tether length is shown to reduce the activation energies following a power-law decay to at least 9.2 kcal/mol for DAECn=8, though this convergence in activation free energy for barrier is attained by DAECn=5 (ΔG‡DMF = 10.9 kcal/mol). The impact different ring substituents and tether functional groups have on the rate of activation energy decay, and whether a power-law decay functional form is ubiquitous are intriguing avenues for future research. Synthetic efforts utilizing an Ullmann coupling or SNAr as a key step show promise and with additional optimization would lead to experimental measurement of these conformational barriers. A complete mapping of the conformational ensemble would be useful but is likely only worth pursuing with assistance from automated transition state search techniques [23,31,32,33,34]. In understanding the relationship between intramolecular tether length and DAEC racemization, this information may provide guidance toward future synthesis of diarylether ligands or pharmaceuticals with atropisomeric properties created or tuned by the attachment of an intramolecular tether.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/sym13112127/s1. Figure S1: Plots of the relative activation energies for isomerization between enantiomeric conformations of DAEC of varying tether lengths (6 + n carbons) within an implicit solvent of benzene and dimethylformamide; and Cartesian coordinates of optimized stationary points.
Author Contributions
Conceptualization, N.J.D. and K.A.M.; methodology, N.J.D. and K.A.M.; software, T.J.S. and N.J.D.; validation, T.M.O. and Z.T.; formal analysis, T.J.S.; investigation, T.J.S., H.T., T.M.O. and Z.T.; resources, N.J.D. and K.A.M.; data curation, T.J.S., H.T., T.M.O. and Z.T.; writing—original draft preparation, T.J.S. and K.A.M.; writing—review and editing, T.J.S., K.A.M. and N.J.D.; visualization, T.J.S. and K.A.M.; supervision, N.J.D. and K.A.M.; project administration, T.J.S., N.J.D. and K.A.M.; funding acquisition, T.J.S., N.J.D. and K.A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by start-up funding from the University of Memphis Department of Chemistry, National Science Foundation (NSF) GRFP-1451514 (for T.J.S.), American Chemical Society Petroleum Research Fund 56502-UR1 (for K.A.M.), and NSF CAREER BIO-1846408 (for N.J.D.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the above Supplementary Materials.
Acknowledgments
The authors thank the High Performance Computing Center and the Computational Research on Materials Institute at the University of Memphis (CROMIUM) for providing generous resources for this research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brooks, W.H.; Guida, W.C.; Daniel, K.G. The Significance of Chirality in Drug Design and Development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Ōki, M. Recent Advances in Atropisomerism. In Topics in Stereochemistry; John Wiley & Sons: Hoboken, NJ, USA, 1983. [Google Scholar] [CrossRef]
- Glunz, P.W. Recent encounters with atropisomerism in drug discovery. Bioorg. Med. Chem. Lett. 2018, 28, 53–60. [Google Scholar] [CrossRef] [PubMed]
- LaPlante, S.R.; Edwards, P.J.; Fader, L.D.; Jakalian, A.; Hucke, O. Revealing Atropisomer Axial Chirality in Drug Discovery. ChemMedChem 2011, 6, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Panossian, A.; Leroux, F.R.; Colobert, F. Recent advances and new concepts for the synthesis of axially stereoenriched biaryls. Chem. Soc. Rev. 2015, 44, 3418–3430. [Google Scholar] [CrossRef] [Green Version]
- Smyth, J.E.; Butler, N.M.; Keller, P.A. A twist of nature—The significance of atropisomers in biological systems. Nat. Prod. Rep. 2015, 32, 1562–1583. [Google Scholar] [CrossRef] [Green Version]
- Bringmann, G.; Gulder, T.; Gulder, T.A.M.; Breuning, M. Atroposelective Total Synthesis of Axially Chiral Biaryl Natural Products. Chem. Rev. 2010, 111, 563–639. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, H.; Fujita, Y.; Banno, N.; Watanabe, K.; Kimura, Y.; Manosroi, A.; Manosroi, J.; Akihisa, T. Three new cyclic diarylheptanoids and other phenolic compounds from the bark of Myrica rubra and their melanogenesis inhibitory and radical scavenging activities. J. Oleo Sci. 2010, 59, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salih, M.Q.; Beaudry, C.M. Chirality in Diarylether Heptanoids: Synthesis of Myricatomentogenin, Jugcathanin and Congeners. Org. Lett. 2012, 14, 4026–4029. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Morikawa, T.; Toguchida, I.; Ando, S.; Matsuda, H.; Yoshikawa, M. Inhibitors of nitric oxide production from the bark of Myrica rubra: Structures of new biphenyl type diarylheptanoid glycosides and taraxerane type triterpene. Bioorg. Med. Chem. 2002, 10, 4005–4012. [Google Scholar] [CrossRef]
- Hochmuth, D.H.; König, W.A. Synthesis, resolution and determination of energy barriers to rotation of atropisomeric, planar-chiral [n]paracyclophanes by dynamic enantioselective gas chromatography and computer simulation. Tetrahedron Asymmetry 1999, 10, 1089–1097. [Google Scholar] [CrossRef]
- Kotha, S.; Shirbhate, M.E.; Waghule, G.T. Selected synthetic strategies to cyclophanes. Beilstein J. Org. Chem. 2015, 11, 1274–1331. [Google Scholar] [CrossRef]
- Ueda, T.; Kanomata, N.; Machida, H. Synthesis of Planar-Chiral Paracyclophanes via Samarium (II)-Catalyzed Intramolecular Pinacol Coupling. Org. Lett. 2005, 7, 2365–2368. [Google Scholar] [CrossRef] [PubMed]
- Kanda, K.; Hamanaka, R.; Endo, K.; Shibata, T. Asymmetric ortho-lithiation of 1,n-dioxa[n]paracyclophane derivatives for the generation of planar chirality. Tetrahedron 2012, 68, 1407–1416. [Google Scholar] [CrossRef]
- Scharwächter, K.P.; Hochmuth, D.H.; Dittmann, H.; König, W.A. Synthesis, resolution, and investigation of the rotational interconversion process of atropisomeric 1,n-diaza[n]paracyclophanes using cyclodextrin-mediated capillary zone electrophoresis. Chirality 2001, 13, 679–690. [Google Scholar] [CrossRef]
- Ferguson, D.M.; Raber, D.J. A new approach to probing conformational space with molecular mechanics: Random incremental pulse search. J. Am. Chem. Soc. 1989, 111, 4371–4378. [Google Scholar] [CrossRef]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE); Chemical Computing Group ULC: Montreal, QC, Canada, 2021. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Pattawong, O.; Salih, M.Q.; Rosson, N.T.; Beaudry, C.M.; Cheong, P.H.-Y. The nature of persistent conformational chirality, racemization mechanisms, and predictions in diarylether heptanoid cyclophane natural products. Org. Biomol. Chem. 2014, 12, 3303–3309. [Google Scholar] [CrossRef]
- Zhu, Z.-Q.; Salih, M.Q.; Fynn, E.; Bain, A.D.; Beaudry, C.M. The Garuganin and Garugamblin Diarylether Heptanoids: Total Synthesis and Determination of Chiral Properties Using Dynamic NMR. J. Org. Chem. 2013, 78, 2881–2896. [Google Scholar] [CrossRef]
- Gonzalez, G.I.; Zhu, J. A Unified Strategy toward the Synthesis of Acerogenin-Type Macrocycles: Total Syntheses of Acerogenins A, B, C, and L and Aceroside IV. J. Org. Chem. 1999, 64, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Atkinson, J. Synthesis of Phytyl- and Chroman-Derivatized Photoaffinity Labels Based on α-Tocopherol. J. Org. Chem. 2000, 65, 2560–2567. [Google Scholar] [CrossRef] [PubMed]
- More, J.D.; Finney, N.S. A Simple and Advantageous Protocol for the Oxidation of Alcohols with o-Iodoxybenzoic Acid (IBX). Org. Lett. 2002, 4, 3001–3003. [Google Scholar] [CrossRef] [PubMed]
- Burgos, C.H.; Barder, T.E.; Huang, X.; Buchwald, S.L. Significantly Improved Method for the Pd-Catalyzed Coupling of Phenols with Aryl Halides: Understanding Ligand Effects. Angew. Chem. Int. Ed. 2006, 45, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Jourdant, A.; González-Zamora, E.; Zhu, J. Wilkinson’s Catalyst Catalyzed Selective Hydrogenation of Olefin in the Presence of an Aromatic Nitro Function: A Remarkable Solvent Effect. J. Org. Chem. 2002, 67, 3163–3164. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Ren, J.; Zeng, B.-B. Oxidation of aromatic amines into nitroarenes with m-CPBA. Tetrahedron Lett. 2014, 55, 1581–1584. [Google Scholar] [CrossRef]
- Guan, Y.; Ingman, V.M.; Rooks, B.J.; Wheeler, S.E. AARON: An Automated Reaction Optimizer for New Catalysts. J. Chem. Theory Comput. 2018, 14, 5249–5261. [Google Scholar] [CrossRef]
- Jacobson, L.D.; Bochevarov, A.D.; Watson, M.A.; Hughes, T.F.; Rinaldo, D.; Ehrlich, S.; Steinbrecher, T.B.; Vaitheeswaran, S.; Philipp, D.M.; Halls, M.D.; et al. Automated Transition State Search and Its Application to Diverse Types of Organic Reactions. J. Chem. Theory Comput. 2017, 13, 5780–5797. [Google Scholar] [CrossRef]
- Robertson, C.; Habershon, S. Simple position and orientation preconditioning scheme for minimum energy path calculations. J. Comput. Chem. 2021, 42, 761–770. [Google Scholar] [CrossRef]
- Maeda, S.; Harabuchi, Y.; Ono, Y.; Taketsugu, T.; Morokuma, K. Intrinsic reaction coordinate: Calculation, bifurcation, and automated search. Int. J. Quantum Chem. 2014, 115, 258–269. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).