
Organ-Specific Mitochondrial Alterations Following Ischemia–Reperfusion Injury in Post-Cardiac Arrest Syndrome: A Comprehensive Review

 , , , , and
, , , , and 
{kind=link}
Abstract
1. Introduction
2. The Pivotal Role of Mitochondria in Cellular Bioenergetics and Homeostasis
3. Organ-Specific Metabolism during Ischemia and Reperfusion
3.1. Brain Metabolism and IR Injury
3.2. Heart Metabolism and IR Injury
3.3. Kidney Metabolism and IR Injury
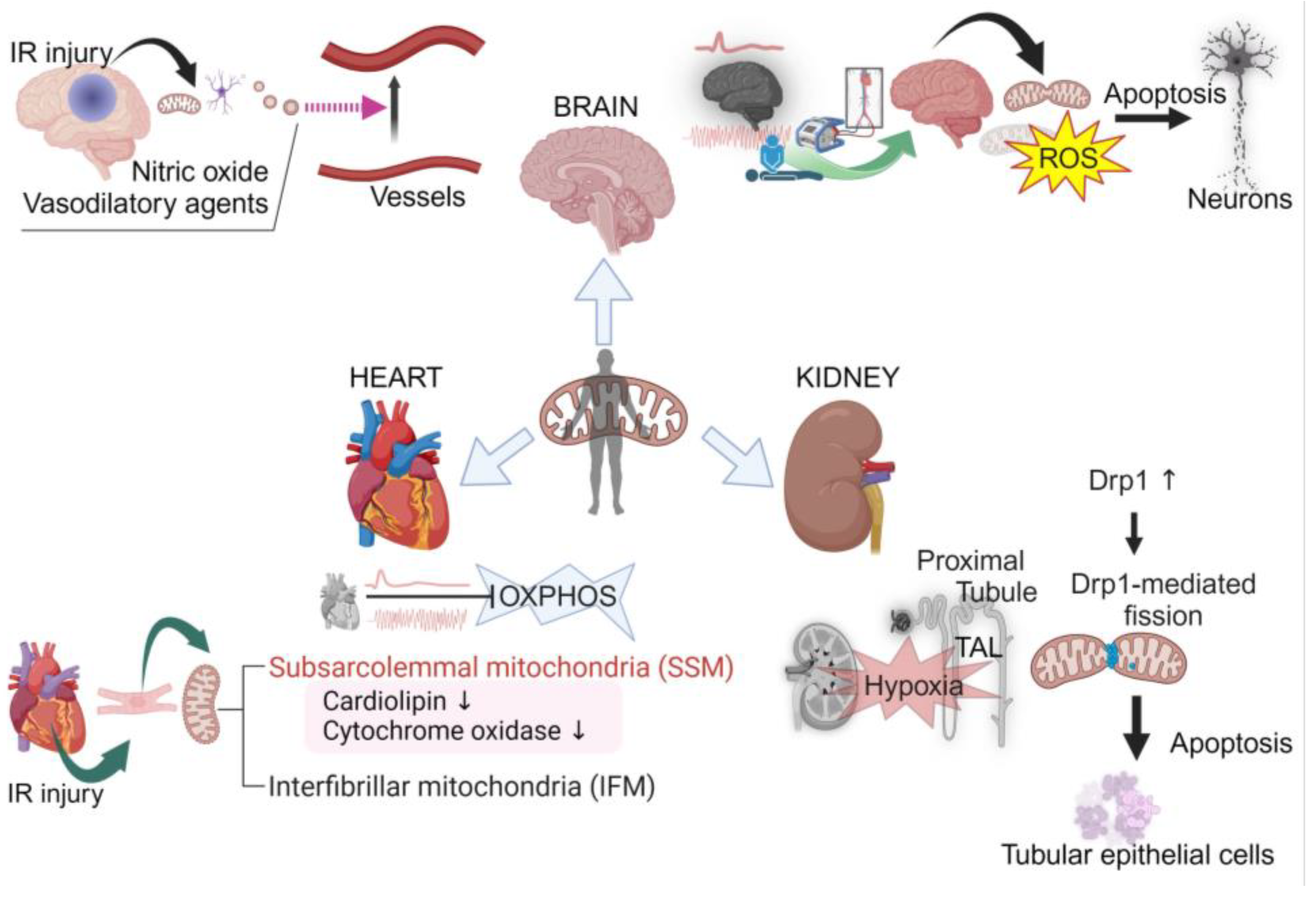
4. Organ-Specific Mitochondrial Responses to Ischemia–Reperfusion Injury
4.1. Brain Mitochondrial Response and Vulnerability in IR Injury
4.2. Heterogeneity of Cardiac Mitochondria and Their Role in IR Injury
4.3. Renal Mitochondrial Resilience and Vulnerability in Ischemia–Reperfusion Injury
5. Comparison of Mitochondrial Responses and Dynamics to Ischemia–Reperfusion Injury between the Brain, Heart, and Kidneys
6. Pharmaceutical Approaches in Ischemia–Reperfusion Injury with Targeted Mitochondrial Protection
7. Mitochondrial Transplantation as an Emerging Strategy for IR Injury
8. Future Directions
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IR: | ischemia–reperfusion |
| IMM: | inner mitochondrial membrane |
| ETC: | electron transport chain |
| ROS: | reactive oxygen species |
| ATP: | adenosine triphosphate |
| MMP: | mitochondrial membrane potential |
| mPTP: | mitochondrial permeability transition pore |
| OXPHOS: | oxidative phosphorylation |
| MPT: | mitochondrial permeability transition |
| SSM: | subsarcolemmal mitochondria |
| IFM: | interfibrillar mitochondria |
| PNM: | perinuclear mitochondria |
| TAL: | thick ascending limbs |
| Drp1: | dynamin-related protein 1 |
| AKI: | acute kidney injury |
| BNIP3: | BCL2/adenovirus E1B 19kDa interacting protein 3 |
| PINK1: | PTEN-induced putative kinase 1 |
| MTx: | mitochondrial transplantation |
References
- Neumar, R.W.; Nolan, J.P.; Adrie, C.; Aibiki, M.; Berg, R.A.; Bottiger, B.W.; Callaway, C.; Clark, R.S.; Geocadin, R.G.; Jauch, E.C.; et al. Post-cardiac arrest syndrome: Epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation 2008, 118, 2452–2483. [Google Scholar] [PubMed]
- Callaway, C.W.; Donnino, M.W.; Fink, E.L.; Geocadin, R.G.; Golan, E.; Kern, K.B.; Leary, M.; Meurer, W.J.; Peberdy, M.A.; Thompson, T.M.; et al. Part 8: Post-Cardiac Arrest Care: 2015 American Heart Association Guidelines Update for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2015, 132 (Suppl. 2), S465–S482. [Google Scholar] [CrossRef] [PubMed]
- Kilbaugh, T.J.; Sutton, R.M.; Karlsson, M.; Hansson, M.J.; Naim, M.Y.; Morgan, R.W.; Bratinov, G.; Lampe, J.W.; Nadkarni, V.M.; Becker, L.B.; et al. Persistently Altered Brain Mitochondrial Bioenergetics After Apparently Successful Resuscitation From Cardiac Arrest. J. Am. Heart Assoc. 2015, 4, e002232. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, I.M.; Radhakrishnan, J.; Gazmuri, R.J. Targeting mitochondria for resuscitation from cardiac arrest. Crit. Care Med. 2008, 36 (Suppl. 11), S440–S446. [Google Scholar] [CrossRef]
- Kim, J.; Villarroel, J.P.; Zhang, W.; Yin, T.; Shinozaki, K.; Hong, A.; Lampe, J.W.; Becker, L.B. The Responses of Tissues from the Brain, Heart, Kidney, and Liver to Resuscitation following Prolonged Cardiac Arrest by Examining Mitochondrial Respiration in Rats. Oxid. Med. Cell. Longev. 2016, 2016, 7463407. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.C.; Li, C.Q.; Shao, Z.H.; Hoang, T.; Chan, K.C.; Hamann, K.J.; Becker, L.B.; Vanden Hoek, T.L. Transient and partial mitochondrial inhibition for the treatment of postresuscitation injury: Getting it just right. Crit. Care Med. 2006, 34 (Suppl. 12), S474–S482. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, S.; Stride, N.; Bro-Jeppesen, J.; Holmberg, M.J.; Kjaergaard, J.; Larsen, S.; Donnino, M.W.; Hassager, C.; Dela, F. Mitochondrial dysfunction in adults after out-of-hospital cardiac arrest. Eur. Heart J. Acute Cardiovasc. Care 2020, 9 (Suppl. 4), S138–S144. [Google Scholar] [CrossRef]
- Neumar, R.W. Molecular mechanisms of ischemic neuronal injury. Ann. Emerg. Med. 2000, 36, 483–506. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Dorweiler, B.; Pruefer, D.; Andrasi, T.B.; Maksan, S.M.; Schmiedt, W.; Neufang, A.; Vahl, C.F. Ischemia-Reperfusion Injury: Pathophysiology and Clinical Implications. Eur. J. Trauma Emerg. Surg. 2007, 33, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Singh, N.; Jaggi, A.S.; Maslov, L. Clinical Applicability of Conditioning Techniques in Ischemia-Reperfusion Injury: A Review of the Literature. Curr. Cardiol. Rev. 2021, 17, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Collard, C.D.; Gelman, S. Pathophysiology, clinical manifestations, and prevention of ischemia-reperfusion injury. Anesthesiology 2001, 94, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [PubMed]
- Gunata, M.; Parlakpinar, H. A review of myocardial ischaemia/reperfusion injury: Pathophysiology, experimental models, biomarkers, genetics and pharmacological treatment. Cell Biochem. Funct. 2021, 39, 190–217. [Google Scholar] [CrossRef] [PubMed]
- San-Millan, I. The Key Role of Mitochondrial Function in Health and Disease. Antioxidants 2023, 12, 782. [Google Scholar] [CrossRef]
- Niizuma, K.; Yoshioka, H.; Chen, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Okami, N.; Chan, P.H. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta 2010, 1802, 92–99. [Google Scholar] [CrossRef]
- Marin, W.; Marin, D.; Ao, X.; Liu, Y. Mitochondria as a therapeutic target for cardiac ischemia-reperfusion injury (Review). Int. J. Mol. Med. 2021, 47, 485–499. [Google Scholar] [CrossRef]
- Sims, N.R.; Anderson, M.F. Mitochondrial contributions to tissue damage in stroke. Neurochem. Int. 2002, 40, 511–526. [Google Scholar] [CrossRef]
- Jassem, W.; Heaton, N.D. The role of mitochondria in ischemia/reperfusion injury in organ transplantation. Kidney Int. 2004, 66, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Huttemann, M. Molecular mechanisms of ischemia-reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef]
- Averbeck, D.; Rodriguez-Lafrasse, C. Role of Mitochondria in Radiation Responses: Epigenetic, Metabolic, and Signaling Impacts. Int. J. Mol. Sci. 2021, 22, 11047. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M., 3rd; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015, 25, 158–170. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.P.; Kohler, M.; Graff, C.; Oldfors, A.; Magnuson, M.A.; Berggren, P.O.; Larsson, N.G. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat. Genet. 2000, 26, 336–340. [Google Scholar] [CrossRef]
- Valenti, D.; Atlante, A. Mitochondrial Bioenergetics in Different Pathophysiological Conditions. Int. J. Mol. Sci. 2021, 22, 7562. [Google Scholar] [CrossRef] [PubMed]
- van der Laan, M.; Bohnert, M.; Wiedemann, N.; Pfanner, N. Role of MINOS in mitochondrial membrane architecture and biogenesis. Trends Cell Biol. 2012, 22, 185–192. [Google Scholar] [CrossRef]
- Mannella, C.A.; Lederer, W.J.; Jafri, M.S. The connection between inner membrane topology and mitochondrial function. J. Mol. Cell Cardiol. 2013, 62, 51–57. [Google Scholar] [CrossRef]
- Rampelt, H.; Zerbes, R.M.; van der Laan, M.; Pfanner, N. Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Wollweber, F.; von der Malsburg, K.; van der Laan, M. Mitochondrial contact site and cristae organizing system: A central player in membrane shaping and crosstalk. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1481–1489. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox. Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, C.; Feng, C.; Yan, C.; Yu, Y.; Chen, Z.; Guo, C.; Wang, X. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox. Rep. 2022, 27, 45–52. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants 2022, 11, 1995. [Google Scholar] [CrossRef]
- Duszynski, J.; Koziel, R.; Brutkowski, W.; Szczepanowska, J.; Zablocki, K. The regulatory role of mitochondria in capacitative calcium entry. Biochim. Biophys. Acta 2006, 1757, 380–387. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Vegas, A.; Eisner, V.; Jaimovich, E. Skeletal muscle excitation-metabolism coupling. Arch. Biochem. Biophys. 2019, 664, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [PubMed]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Vorobjev, I.A.; Popkov, V.A.; Babenko, V.A.; Zorova, L.D.; Pevzner, I.B.; Silachev, D.N.; Zorov, S.D.; Andrianova, N.V.; Plotnikov, E.Y. Lessons from the Discovery of Mitochondrial Fragmentation (Fission): A Review and Update. Cells 2019, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef]
- Cojocaru, K.A.; Luchian, I.; Goriuc, A.; Antoci, L.M.; Ciobanu, C.G.; Popescu, R.; Vlad, C.E.; Blaj, M.; Foia, L.G. Mitochondrial Dysfunction, Oxidative Stress, and Therapeutic Strategies in Diabetes, Obesity, and Cardiovascular Disease. Antioxidants 2023, 12, 658. [Google Scholar] [CrossRef]
- Onishi, M.; Okamoto, K. Mitochondrial clearance: Mechanisms and roles in cellular fitness. FEBS Lett. 2021, 595, 1239–1263. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef] [PubMed]
- Siesjo, B.K.; Plum, F. Cerebral energy metabolism in normoxia and in hypoxia. Acta Anaesthesiol. Scand. Suppl. 1971, 45, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Siesjo, B.K. Cell damage in the brain: A speculative synthesis. J. Cereb. Blood Flow Metab. 1981, 1, 155–185. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Duffy, T.E. Cerebral energy metabolism during transient ischemia and recovery in the gerbil. J. Neurochem. 1977, 28, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. What makes the mitochondria a killer? Can we condition them to be less destructive? Biochim. Biophys. Acta 2011, 1813, 1302–1308. [Google Scholar] [CrossRef]
- Duchen, M.R.; McGuinness, O.; Brown, L.A.; Crompton, M. On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc. Res. 1993, 27, 1790–1794. [Google Scholar] [CrossRef]
- Chen, Q.; Younus, M.; Thompson, J.; Hu, Y.; Hollander, J.M.; Lesnefsky, E.J. Intermediary metabolism and fatty acid oxidation: Novel targets of electron transport chain-driven injury during ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H787–H795. [Google Scholar] [CrossRef]
- Faivre, A.; Verissimo, T.; Auwerx, H.; Legouis, D.; de Seigneux, S. Tubular Cell Glucose Metabolism Shift During Acute and Chronic Injuries. Front. Med. 2021, 8, 742072. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, T.A.; Gustafsson, H.; Palm, F. Kidney outer medulla mitochondria are more efficient compared with cortex mitochondria as a strategy to sustain ATP production in a suboptimal environment. Am. J. Physiol. Renal Physiol. 2018, 315, F677–F681. [Google Scholar] [CrossRef] [PubMed]
- Loor, G.; Kondapalli, J.; Iwase, H.; Chandel, N.S.; Waypa, G.B.; Guzy, R.D.; Vanden Hoek, T.L.; Schumacker, P.T. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim. Biophys. Acta 2011, 1813, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Seager, R.; Lee, L.; Henley, J.M.; Wilkinson, K.A. Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal. 2020, 4, NS20200008. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, V.; Lauterbach, M.; Schuman, E.M. Spatially Stable Mitochondrial Compartments Fuel Local Translation during Plasticity. Cell 2019, 176, 73–84.e15. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.T.; Honick, A.S.; Reynolds, I.J. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci. 2006, 26, 7035–7045. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.L., Jr.; Kwon, S.K.; Lee, A.; Shaw, R.; Polleux, F. MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat. Commun. 2018, 9, 5008. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, T.; Pendin, D.; Montagna, A.; Bova, S.; Ghelli, A.M.; Daga, A. Manipulation of Mitochondria Dynamics Reveals Separate Roles for Form and Function in Mitochondria Distribution. Cell Rep. 2018, 23, 1742–1753. [Google Scholar] [CrossRef]
- Fox, P.T.; Raichle, M.E.; Mintun, M.A.; Dence, C. Nonoxidative glucose consumption during focal physiologic neural activity. Science 1988, 241, 462–464. [Google Scholar] [CrossRef]
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Macaskill, A.F.; Rinholm, J.E.; Twelvetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; Aspenstrom, P.; Attwell, D.; Kittler, J.T. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 2009, 61, 541–555. [Google Scholar] [CrossRef]
- Vaccaro, V.; Devine, M.J.; Higgs, N.F.; Kittler, J.T. Miro1-dependent mitochondrial positioning drives the rescaling of presynaptic Ca2+ signals during homeostatic plasticity. EMBO Rep. 2017, 18, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P. Mitochondrial bioenergetics and neurodegeneration: A paso doble. Neural Regen. Res. 2021, 16, 686–687. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.J.; Pekkurnaz, G. Powerhouse of the mind: Mitochondrial plasticity at the synapse. Curr. Opin. Neurobiol. 2019, 57, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Faria-Pereira, A.; Morais, V.A. Synapses: The Brain’s Energy-Demanding Sites. Int. J. Mol. Sci. 2022, 23, 3627. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.Y.; Sheng, Z.H. Regulation of mitochondrial transport in neurons. Exp. Cell Res. 2015, 334, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, R.; Garcia, G.C.; Bartol, T.M.; Lee, C.T.; Khandelwal, P.; Liu, E.; Spencer, D.J.; Husar, A.; Bushong, E.A.; Phan, S.; et al. Morphological principles of neuronal mitochondria. J. Comp. Neurol. 2022, 530, 886–902. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E. A shape shifting organelle: Unusual mitochondrial phenotype determined with three-dimensional electron microscopy reconstruction. Neural Regen. Res. 2016, 11, 900–901. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Fiskum, G.; Danilov, C.A.; Mehrabian, Z.; Bambrick, L.L.; Kristian, T.; McKenna, M.C.; Hopkins, I.; Richards, E.M.; Rosenthal, R.E. Postischemic oxidative stress promotes mitochondrial metabolic failure in neurons and astrocytes. Ann. N. Y. Acad. Sci. 2008, 1147, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Lu, M.H.; Yuan, D.J.; Xu, D.E.; Yao, P.P.; Ji, W.L.; Chen, H.; Liu, W.L.; Yan, C.X.; Xia, Y.Y.; et al. Mitochondrial Dysfunction in Neural Injury. Front. Neurosci. 2019, 13, 30. [Google Scholar] [CrossRef]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar]
- Robichaux, D.J.; Harata, M.; Murphy, E.; Karch, J. Mitochondrial permeability transition pore-dependent necrosis. J. Mol. Cell Cardiol. 2023, 174, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Fels, J.A.; Manfredi, G. Sex Differences in Ischemia/Reperfusion Injury: The Role of Mitochondrial Permeability Transition. Neurochem. Res. 2019, 44, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zeng, W.; Zhang, Y.; Yu, Q.; Zeng, M.; Gan, J.; Zhang, W.; Jiang, X.; Li, H. Focus on the role of mitochondria in NLRP3 inflammasome activation: A prospective target for the treatment of ischemic stroke (Review). Int. J. Mol. Med. 2022, 49, 74. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Kaindl, A.M. Neuronal death and oxidative stress in the developing brain. Antioxid. Redox Signal. 2011, 14, 1535–1550. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Centonze, D.; Bernardi, G. Cellular factors controlling neuronal vulnerability in the brain: A lesson from the striatum. Neurology 2000, 55, 1249–1255. [Google Scholar] [CrossRef]
- Zhao, N.; Zhuo, X.; Lu, Y.; Dong, Y.; Ahmed, M.E.; Tucker, D.; Scott, E.L.; Zhang, Q. Intranasal Delivery of a Caspase-1 Inhibitor in the Treatment of Global Cerebral Ischemia. Mol. Neurobiol. 2017, 54, 4936–4952. [Google Scholar] [CrossRef]
- Busija, D.W.; Rutkai, I.; Dutta, S.; Katakam, P.V. Role of Mitochondria in Cerebral Vascular Function: Energy Production, Cellular Protection, and Regulation of Vascular Tone. Compr. Physiol. 2016, 6, 1529–1548. [Google Scholar] [PubMed]
- Christie, I.N.; Theparambil, S.M.; Braga, A.; Doronin, M.; Hosford, P.S.; Brazhe, A.; Mascarenhas, A.; Nizari, S.; Hadjihambi, A.; Wells, J.A.; et al. Astrocytes produce nitric oxide via nitrite reduction in mitochondria to regulate cerebral blood flow during brain hypoxia. Cell Rep. 2023, 42, 113514. [Google Scholar] [CrossRef] [PubMed]
- White, B.C.; Grossman, L.I.; O’Neil, B.J.; DeGracia, D.J.; Neumar, R.W.; Rafols, J.A.; Krause, G.S. Global brain ischemia and reperfusion. Ann. Emerg. Med. 1996, 27, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Vongsfak, J.; Pratchayasakul, W.; Apaijai, N.; Vaniyapong, T.; Chattipakorn, N.; Chattipakorn, S.C. The Alterations in Mitochondrial Dynamics Following Cerebral Ischemia/Reperfusion Injury. Antioxidants 2021, 10, 1384. [Google Scholar] [CrossRef]
- Kumar, R.; Bukowski, M.J.; Wider, J.M.; Reynolds, C.A.; Calo, L.; Lepore, B.; Tousignant, R.; Jones, M.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics following global cerebral ischemia. Mol. Cell Neurosci. 2016, 76, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tian, F.; Kurata, T.; Morimoto, N.; Abe, K. Dynamic changes of mitochondrial fusion and fission proteins after transient cerebral ischemia in mice. J. Neurosci. Res. 2012, 90, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Bakthavachalam, P.; Shanmugam, P.S.T. Mitochondrial dysfunction—Silent killer in cerebral ischemia. J. Neurol. Sci. 2017, 375, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Bruzzese, M.; Chou, S.H.; Ning, M.; Ji, X.; Lo, E.H. Extracellular Mitochondria for Therapy and Diagnosis in Acute Central Nervous System Injury. JAMA Neurol. 2018, 75, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Riva, A.; Tandler, B.; Loffredo, F.; Vazquez, E.; Hoppel, C. Structural differences in two biochemically defined populations of cardiac mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H868–H872. [Google Scholar] [CrossRef]
- Palmer, J.W.; Tandler, B.; Hoppel, C.L. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 1977, 252, 8731–8739. [Google Scholar] [CrossRef] [PubMed]
- Hollander, J.M.; Thapa, D.; Shepherd, D.L. Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: Influence of cardiac pathologies. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1–H14. [Google Scholar] [CrossRef] [PubMed]
- Dabkowski, E.R.; Williamson, C.L.; Bukowski, V.C.; Chapman, R.S.; Leonard, S.S.; Peer, C.J.; Callery, P.S.; Hollander, J.M. Diabetic cardiomyopathy-associated dysfunction in spatially distinct mitochondrial subpopulations. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H359–H369. [Google Scholar] [CrossRef] [PubMed]
- Meerson, F.Z.; Zaletayeva, T.A.; Lagutchev, S.S.; Pshennikova, M.G. Structure and Mass of Mitochondria in the Process of Compensatory Hyperfunction and Hypertrophy of the Heart. Exp. Cell Res. 1964, 36, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Rousou, A.J.; Ericsson, M.; Federman, M.; Levitsky, S.; McCully, J.D. Opening of mitochondrial KATP channels enhances cardioprotection through the modulation of mitochondrial matrix volume, calcium accumulation, and respiration. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1967–H1976. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343. [Google Scholar] [CrossRef]
- Niemann, B.; Schwarzer, M.; Rohrbach, S. Heart and Mitochondria: Pathophysiology and Implications for Cardiac Surgeons. Thorac. Cardiovasc. Surg. 2018, 66, 11–19. [Google Scholar]
- Frank, A.; Bonney, M.; Bonney, S.; Weitzel, L.; Koeppen, M.; Eckle, T. Myocardial ischemia reperfusion injury: From basic science to clinical bedside. Semin. Cardiothorac. Vasc. Anesth. 2012, 16, 123–132. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Tandler, B.; Hoppel, C.L. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 535–565. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Tandler, B.; Ye, J.; Slabe, T.J.; Turkaly, J.; Hoppel, C.L. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am. J. Physiol. 1997, 273 Pt 2, H1544–H1554. [Google Scholar] [CrossRef]
- Ueta, H.; Ogura, R.; Sugiyama, M.; Kagiyama, A.; Shin, G. O2− spin trapping on cardiac submitochondrial particles isolated from ischemic and non-ischemic myocardium. J. Mol. Cell Cardiol. 1990, 22, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Slabe, T.J.; Stoll, M.S.; Minkler, P.E.; Hoppel, C.L. Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2770–H2778. [Google Scholar] [CrossRef] [PubMed]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Lin, P.H. Mitochondria Damage and Kidney Disease. Adv. Exp. Med. Biol. 2017, 982, 529–551. [Google Scholar]
- Nourbakhsh, N.; Singh, P. Role of renal oxygenation and mitochondrial function in the pathophysiology of acute kidney injury. Nephron Clin. Pract. 2014, 127, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Kuang, L.; Hong, C.; Xiang, X.; Liu, J.; Li, Q.; Peng, X.; Zhou, Y.; Wang, H.; Liu, L.; et al. Mitochondrial Drp1 recognizes and induces excessive mPTP opening after hypoxia through BAX-PiC and LRRK2-HK2. Cell Death Dis. 2021, 12, 1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, M.; Xiong, L.; Fan, J.; Zhou, Y.; Li, H.; Peng, X.; Zhong, Z.; Wang, Y.; Huang, F.; et al. Drp1-mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell Death Dis. 2020, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Perry, H.M.; Huang, L.; Wilson, R.J.; Bajwa, A.; Sesaki, H.; Yan, Z.; Rosin, D.L.; Kashatus, D.F.; Okusa, M.D. Dynamin-Related Protein 1 Deficiency Promotes Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 194–206. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Huang, H.; van Dullemen, L.F.A.; Akhtar, M.Z.; Faro, M.L.; Yu, Z.; Valli, A.; Dona, A.; Thezenas, M.L.; Charles, P.D.; Fischer, R.; et al. Proteo-metabolomics reveals compensation between ischemic and non-injured contralateral kidneys after reperfusion. Sci. Rep. 2018, 8, 8539. [Google Scholar] [CrossRef]
- Chatauret, N.; Badet, L.; Barrou, B.; Hauet, T. Ischemia-reperfusion: From cell biology to acute kidney injury. Prog. Urol. 2014, 24 (Suppl. 1), S4–S12. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Yoder, M.C. Renal endothelial dysfunction in acute kidney ischemia reperfusion injury. Cardiovasc. Hematol. Disord. Drug Targets 2014, 14, 3–14. [Google Scholar] [CrossRef]
- Springer, W.; Kahle, P.J. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy 2011, 7, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chen, D.; Si, J.; Hu, Q.; Qin, Z.; Fang, M.; Wang, G. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum. Mol. Genet. 2015, 24, 2528–2538. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, L.; Zhang, Y.; Yu, X.; Sun, X.; Zhu, T.; Li, X.; Liang, W.; Han, Y.; Qin, C. PINK1 Deficiency Ameliorates Cisplatin-Induced Acute Kidney Injury in Rats. Front. Physiol. 2019, 10, 1225. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Zhang, Z.; Dong, Z. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018, 9, 1113. [Google Scholar] [CrossRef]
- Tang, C.; Han, H.; Liu, Z.; Liu, Y.; Yin, L.; Cai, J.; He, L.; Liu, Y.; Chen, G.; Zhang, Z.; et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 2019, 10, 677. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. [Google Scholar] [CrossRef]
- Salvadori, M.; Rosso, G.; Bertoni, E. Update on ischemia-reperfusion injury in kidney transplantation: Pathogenesis and treatment. World J. Transplant. 2015, 5, 52–67. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef]
- Kretzschmar, T.; Wu, J.M.F.; Schulze, P.C. Mitochondrial Homeostasis Mediates Lipotoxicity in the Failing Myocardium. Int. J. Mol. Sci. 2021, 22, 1498. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Asemu, G.; O’Connell, K.A.; Cox, J.W.; Dabkowski, E.R.; Xu, W.; Ribeiro, R.F., Jr.; Shekar, K.C.; Hecker, P.A.; Rastogi, S.; Sabbah, H.N.; et al. Enhanced resistance to permeability transition in interfibrillar cardiac mitochondria in dogs: Effects of aging and long-term aldosterone infusion. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H514–H528. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Bajwa, A. Role of Mitochondrial Therapy for Ischemic-Reperfusion Injury and Acute Kidney Injury. Nephron 2022, 146, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Davani, A.J.; Natarajan, G.K.; Kwok, W.M.; Stowe, D.F.; Camara, A.K.S. Cyclosporin A Increases Mitochondrial Buffering of Calcium: An Additional Mechanism in Delaying Mitochondrial Permeability Transition Pore Opening. Cells 2019, 8, 1052. [Google Scholar] [CrossRef]
- Griffiths, E.J.; Halestrap, A.P. Further evidence that cyclosporin A protects mitochondria from calcium overload by inhibiting a matrix peptidyl-prolyl cis-trans isomerase. Implications for the immunosuppressive and toxic effects of cyclosporin. Biochem. J. 1991, 274 Pt 2, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Huhn, R.; Heinen, A.; Hollmann, M.W.; Schlack, W.; Preckel, B.; Weber, N.C. Cyclosporine A administered during reperfusion fails to restore cardioprotection in prediabetic Zucker obese rats in vivo. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Squadrito, F.; Altavilla, D.; Squadrito, G.; Saitta, A.; Campo, G.M.; Arlotta, M.; Quartarone, C.; Ferlito, M.; Caputi, A.P. Cyclosporin—A reduces leukocyte accumulation and protects against myocardial ischaemia reperfusion injury in rats. Eur. J. Pharmacol. 1999, 364, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Nazareth, W.; Yafei, N.; Crompton, M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin A. J. Mol. Cell Cardiol. 1991, 23, 1351–1354. [Google Scholar] [CrossRef]
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guerin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Wu, D.; Darrah, S.F.; Cheng, F.Y.; Zhao, Z.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Pesonen, U.; Rouru, J.; Huupponen, R.; Koulu, M. Effects of repeated administration of mifepristone and 8-OH-DPAT on expression of preproneuropeptide Y mRNA in the arcuate nucleus of obese Zucker rats. Brain Res. Mol. Brain Res. 1991, 10, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.H.; Ge, J.B.; Li, M.; Xu, H.D.; Wu, F.; Qin, Z.H. Poloxamer 188 protects neurons against ischemia/reperfusion injury through preserving integrity of cell membranes and blood brain barrier. PLoS ONE 2013, 8, e61641. [Google Scholar] [CrossRef]
- Chen, W.N.; Shaikh, M.F.; Bhuvanendran, S.; Date, A.; Ansari, M.T.; Radhakrishnan, A.K.; Othman, I. Poloxamer 188 (P188), A Potential Polymeric Protective Agent for Central Nervous System Disorders: A Systematic Review. Curr. Neuropharmacol. 2022, 20, 799–808. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Cowan, D.B.; Emani, S.M.; Del Nido, P.J. Mitochondrial transplantation: From animal models to clinical use in humans. Mitochondrion 2017, 34, 127–134. [Google Scholar] [CrossRef]
- Hayashida, K.; Takegawa, R.; Shoaib, M.; Aoki, T.; Choudhary, R.C.; Kuschner, C.E.; Nishikimi, M.; Miyara, S.J.; Rolston, D.M.; Guevara, S.; et al. Mitochondrial transplantation therapy for ischemia reperfusion injury: A systematic review of animal and human studies. J. Transl. Med. 2021, 19, 214. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.S.; Parvez, S.; Tabassum, H. Ischemic stroke and mitochondria: Mechanisms and targets. Protoplasma 2020, 257, 335–343. [Google Scholar] [CrossRef]
- Nakamura, Y.; Lo, E.H.; Hayakawa, K. Placental Mitochondria Therapy for Cerebral Ischemia-Reperfusion Injury in Mice. Stroke 2020, 51, 3142–3146. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, Z.; Yan, C.; Pu, K.; Wu, M.; Bai, J.; Li, Y.; Wang, Q. Muscle-derived autologous mitochondrial transplantation: A novel strategy for treating cerebral ischemic injury. Behav. Brain Res. 2019, 356, 322–331. [Google Scholar] [CrossRef]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K.; Takegawa, R.; Endo, Y.; Yin, T.; Choudhary, R.C.; Aoki, T.; Nishikimi, M.; Murao, A.; Nakamura, E.; Shoaib, M.; et al. Exogenous mitochondrial transplantation improves survival and neurological outcomes after resuscitation from cardiac arrest. BMC Med. 2023, 21, 56. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.; Biswas, S.; Afrose, S.; Mita, M.A.; Hasan, M.R.; Shimu, M.S.S.; Paul, G.K.; Chung, S.; Saleh, M.A.; Alshehri, S.; et al. Use of Next-Generation Sequencing for Identifying Mitochondrial Disorders. Curr. Issues Mol. Biol. 2022, 44, 1127–1148. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.J.; Brilhante, V.; Suomalainen, A. Next-generation sequencing for mitochondrial disorders. Br. J. Pharmacol. 2014, 171, 1837–1853. [Google Scholar] [CrossRef]
- Esterhuizen, K.; van der Westhuizen, F.H.; Louw, R. Metabolomics of mitochondrial disease. Mitochondrion 2017, 35, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.A.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020, 34, 428–445. [Google Scholar] [CrossRef]
- Audano, M.; Ferrari, A.; Fiorino, E.; Kuenzl, M.; Caruso, D.; Mitro, N.; Crestani, M.; De Fabiani, E. Energizing Genetics and Epi-genetics: Role in the Regulation of Mitochondrial Function. Curr. Genom. 2014, 15, 436–456. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, E.; Aoki, T.; Endo, Y.; Kazmi, J.; Hagiwara, J.; Kuschner, C.E.; Yin, T.; Kim, J.; Becker, L.B.; Hayashida, K. Organ-Specific Mitochondrial Alterations Following Ischemia–Reperfusion Injury in Post-Cardiac Arrest Syndrome: A Comprehensive Review. Life 2024, 14, 477. https://doi.org/10.3390/life14040477
Nakamura E, Aoki T, Endo Y, Kazmi J, Hagiwara J, Kuschner CE, Yin T, Kim J, Becker LB, Hayashida K. Organ-Specific Mitochondrial Alterations Following Ischemia–Reperfusion Injury in Post-Cardiac Arrest Syndrome: A Comprehensive Review. Life. 2024; 14(4):477. https://doi.org/10.3390/life14040477
Chicago/Turabian StyleNakamura, Eriko, Tomoaki Aoki, Yusuke Endo, Jacob Kazmi, Jun Hagiwara, Cyrus E. Kuschner, Tai Yin, Junhwan Kim, Lance B. Becker, and Kei Hayashida. 2024. "Organ-Specific Mitochondrial Alterations Following Ischemia–Reperfusion Injury in Post-Cardiac Arrest Syndrome: A Comprehensive Review" Life 14, no. 4: 477. https://doi.org/10.3390/life14040477
APA StyleNakamura, E., Aoki, T., Endo, Y., Kazmi, J., Hagiwara, J., Kuschner, C. E., Yin, T., Kim, J., Becker, L. B., & Hayashida, K. (2024). Organ-Specific Mitochondrial Alterations Following Ischemia–Reperfusion Injury in Post-Cardiac Arrest Syndrome: A Comprehensive Review. Life, 14(4), 477. https://doi.org/10.3390/life14040477