Mucopolysaccharidosis Type I

 ,
,  , , ,
, , ,  ,
,  and
and 
Abstract
:1. Introduction
2. Clinical Picture
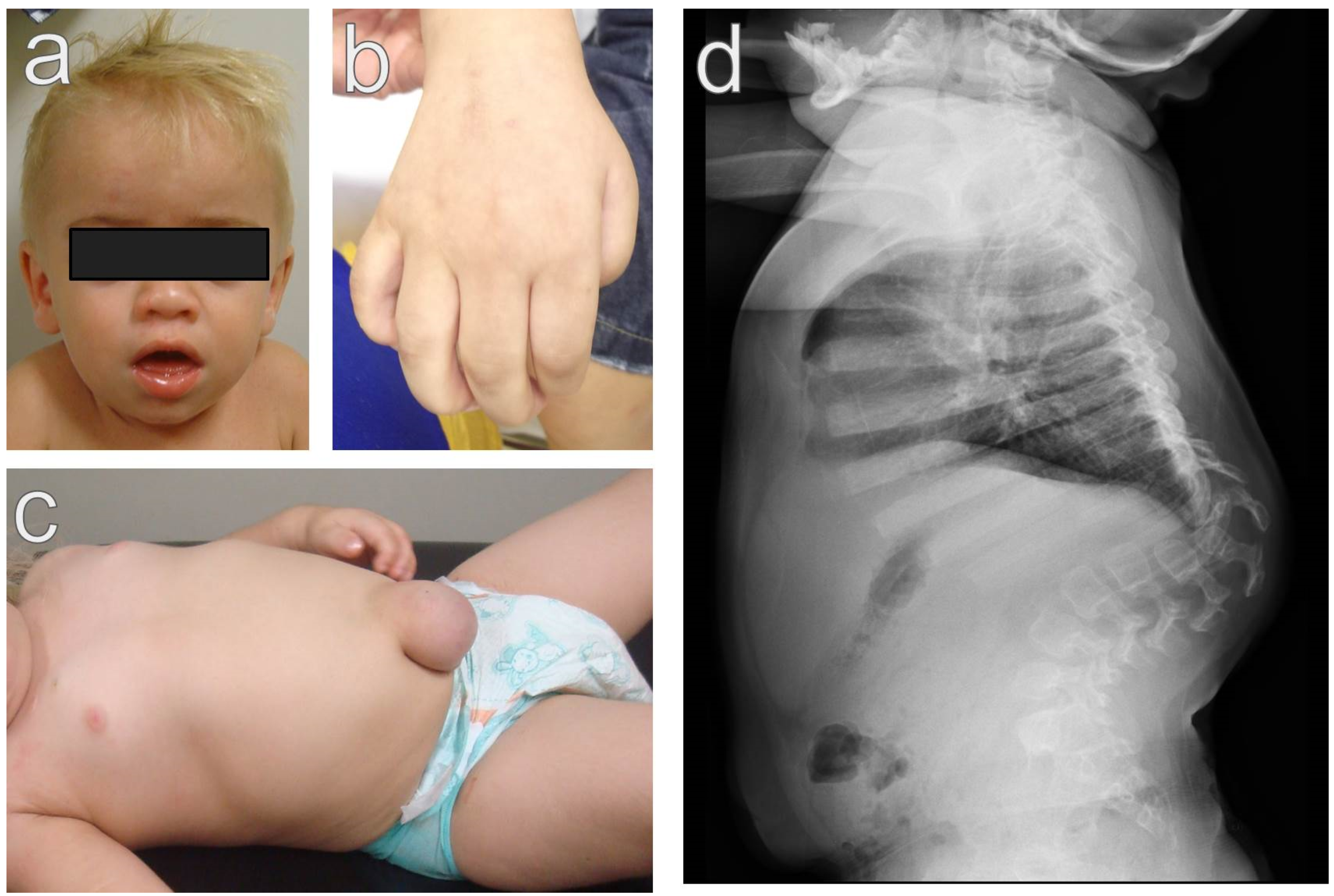
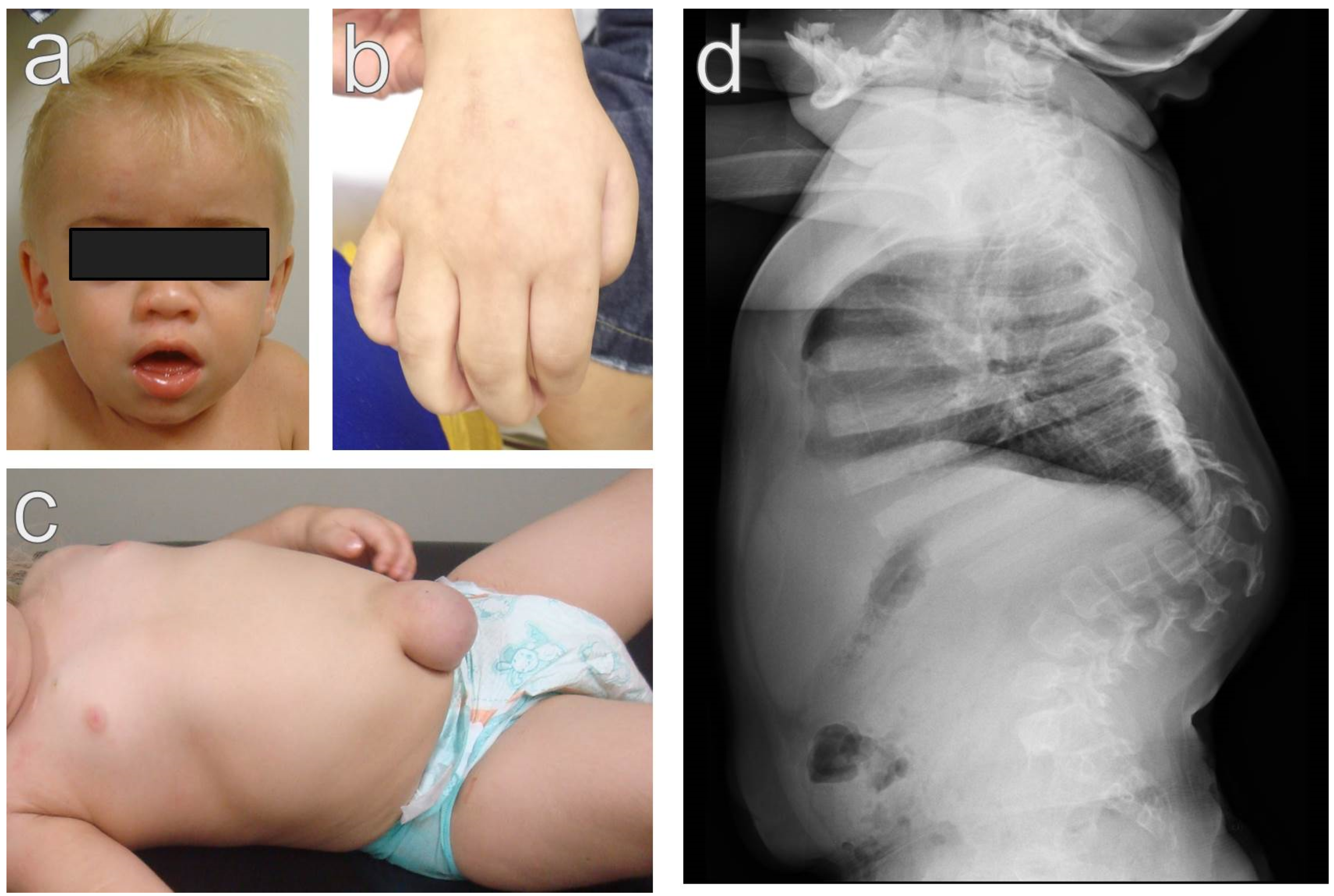
2.1. Severe Phenotype (Hurler Syndrome)
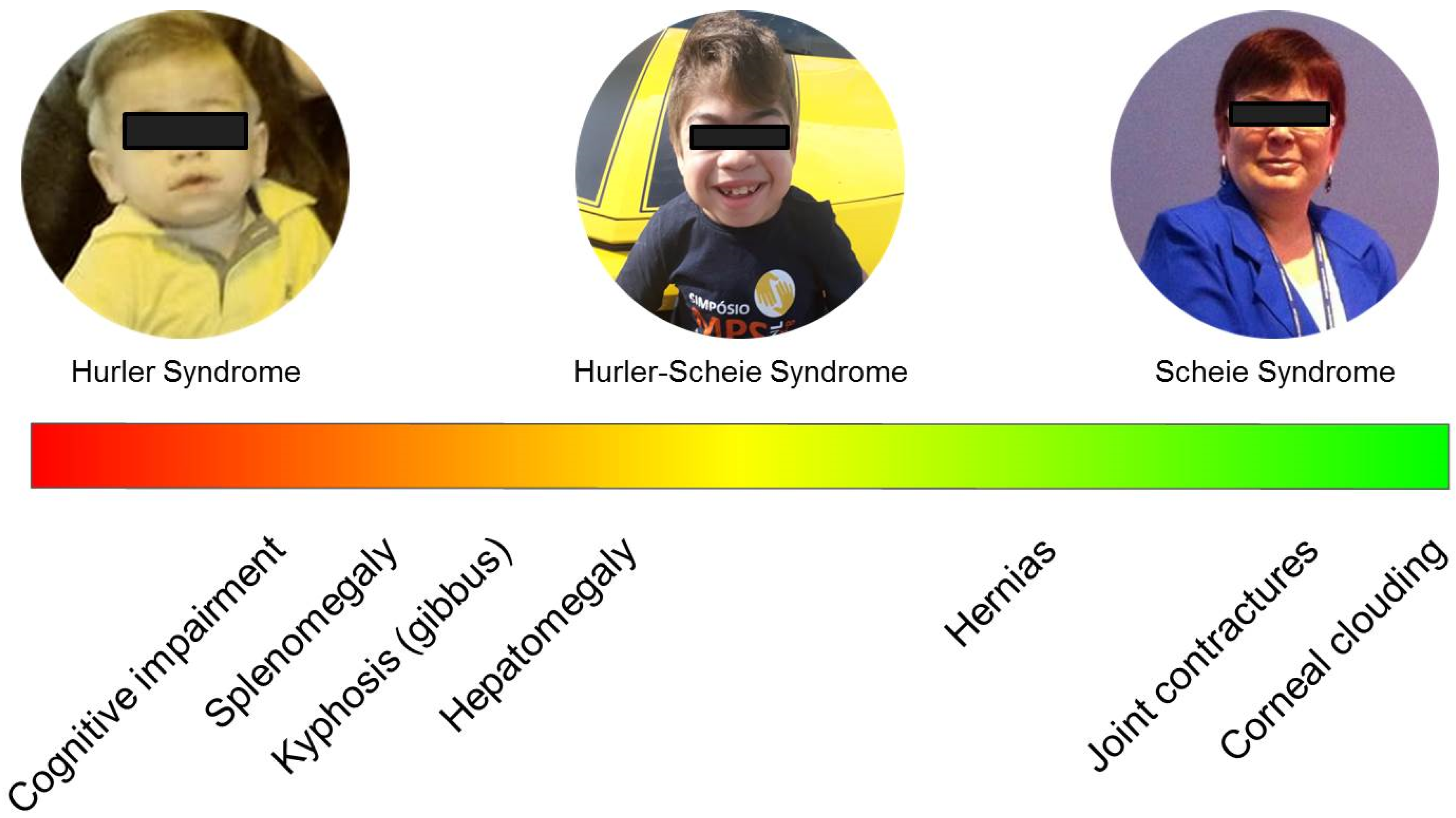
2.2. Attenuated Phenotypes
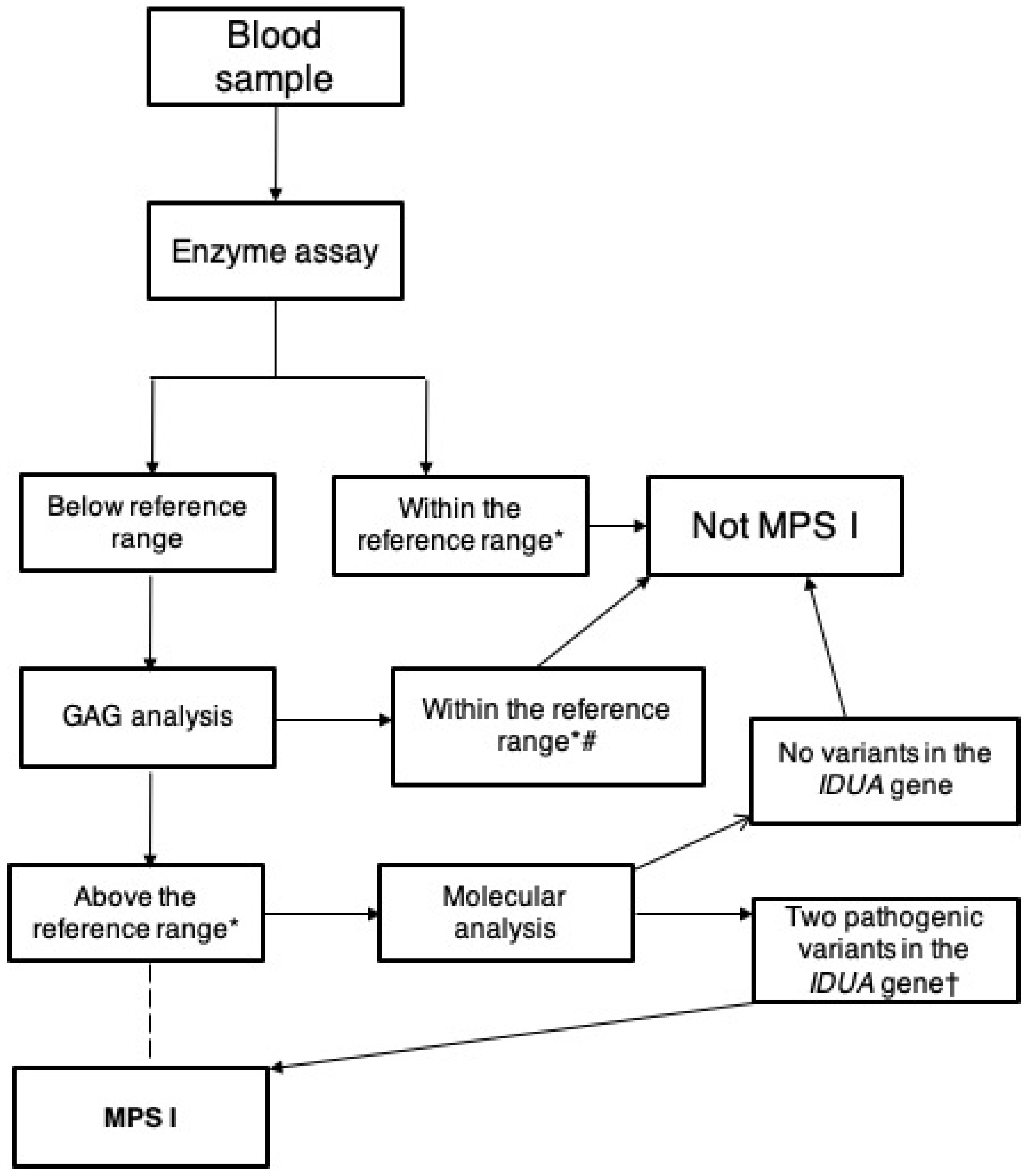
3. Laboratory Diagnosis
3.1. Glycosaminoglycans
3.2. Enzyme Assay
3.3. Molecular Diagnosis
4. Therapeutic Approaches
4.1. Hematopoietic Stem Cell Transplantation
4.2. Intravenous Enzyme Replacement Therapy
4.3. Intravenous Enzyme Replacement Therapy with Fusion Proteins
4.4. Intrathecal Enzyme Replacement Therapy
4.5. Gene Therapy, Gene Editing, and Stop Codon Read Through
4.6. Small Molecules
5. Newborn Screening
6. Genotype–Phenotype Correlation
7. Prospects and Conclusions
Funding
Conflicts of Interest
References
- Giugliani, R. Mucopolysacccharidoses: From understanding to treatment, a century of discoveries. Genet. Mol. Biol. 2012, 35 (Suppl. 1), 924–931. Available online: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S1415-47572012000600006&lng=en&tlng=en (accessed on 13 August 2019). [CrossRef] [PubMed] [Green Version]
- Fratantoni, J.C.; Hall, C.W.; Neufeld, E.F. Hurler and hunter syndromes: Mutual correction of the defect in cultured fibroblasts. Science 1968, 162, 570–572. Available online: http://www.ncbi.nlm.nih.gov/pubmed/4236721 (accessed on 3 September 2019). [CrossRef] [PubMed]
- Tieu, P.T.; Bach, G.; Matynia, A.; Hwang, M.; Neufeld, E.F. Four novel mutations underlying mild or intermediate forms of α-l-iduronidase deficiency (MPS IS and MPS IH/S). Hum. Mutat. 1995, 6, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Sandeep, N.; Rawal, S.; Dabla, S.; Aggarwal, S. Manjri hurler scheie disease. J. Assoc. Physicians India 2015, 63, 83–84. Available online: http://www.ncbi.nlm.nih.gov/pubmed/27608704 (accessed on 14 January 2020). [PubMed]
- Parini, R.; Deodato, F.; Di Rocco, M.; Lanino, E.; Locatelli, F.; Messina, C.; Rovelli, A.; Scarpa, M. Open issues in mucopolysaccharidosis type I-hurler. Orphanet J. Rare Dis. 2017, 12, 112. Available online: http://www.ncbi.nlm.nih.gov/pubmed/28619065 (accessed on 15 April 2018). [CrossRef] [PubMed]
- Beck, M.; Arn, P.; Giugliani, R.; Muenzer, J.; Okuyama, T.; Taylor, J.; Fallet, S. The natural history of MPS I: Global perspectives from the MPS I Registry. Genet. Med. 2014, 16, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Rao, K.S.; Adhikari, S.; Singh, S.; Poudel, S.; Basnet, S.; Bishwakarma, G. Hurler syndrome. J. Nepal Paediatr. Soc. 2016, 36, 295–297. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, E.G.; Nestrasil, I.; Rudser, K.; Delaney, K.; Kovac, V.; Ahmed, A.; Yund, B.; Orchard, P.J.; Eisengart, J.; Niklason, G.R.; et al. Neurocognition across the spectrum of mucopolysaccharidosis type I: Age, severity, and treatment. Mol. Genet. Metab. 2015, 116, 61–68. Available online: http://www.ncbi.nlm.nih.gov/pubmed/26095521 (accessed on 16 April 2018). [CrossRef] [Green Version]
- Hanson, M.; Lupski, J.R.; Hicks, J.; Metry, D. Association of dermal melanocytosis with lysosomal storage disease: Clinical features and hypotheses regarding pathogenesis. Arch. Dermatol. 2003, 139, 916–920. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.J.; Li, Y.W.; Wang, T.R.; Hsu, J.C. Bony changes in common mucopolysaccharidoses. Acta Paediatr. Sin. 1996, 37, 178–184. Available online: http://www.ncbi.nlm.nih.gov/pubmed/8755171 (accessed on 21 October 2019).
- Kennedy, J.; Noel, J.; O’Meara, A.; Kelly, P. Foot and ankle abnormalities in the hurler syndrome: Additions to the phenotype. J. Pediatr. Orthop. 2013, 33, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Różdżyńska-Świątkowska, A.; Jurecka, A.; Żuber, Z.; Tylki-Szymańska, A. Can macrosomia or large for gestational age be predictive of mucopolysaccharidosis type I, II and VI? Pediatr. Neonatol. 2016, 57, 181–187. Available online: http://www.ncbi.nlm.nih.gov/pubmed/26522251 (accessed on 21 October 2019). [CrossRef] [PubMed] [Green Version]
- Gardner, D.G. The oral manifestations of Hurler’s syndrome. Oral Surg. Oral Med. Oral Pathol. 1971, 32, 46–57. Available online: http://www.ncbi.nlm.nih.gov/pubmed/4996613 (accessed on 21 October 2019). [CrossRef]
- McGovern, E.; Owens, L.; Nunn, J.; Bolas, A.; Meara, A.O.; Fleming, P. Oral features and dental health in Hurler Syndrome following hematopoietic stem cell transplantation. Int. J. Paediatr. Dent. 2010, 20, 322–329. [Google Scholar] [CrossRef]
- Rasalkar, D.D.; Chu, W.C.W.; Hui, J.; Chu, C.M.; Paunipagar, B.K.; Li, C.K. Pictorial review of mucopolysaccharidosis with emphasis on MRI features of brain and spine. Br. J. Radiol. 2011, 84, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Leighton, S.E.; Papsin, B.; Vellodi, A.; Dinwiddie, R.; Lane, R. Disordered breathing during sleep in patients with mucopolysaccharidoses. Int. J. Pediatr. Otorhinolaryngol. 2001, 58, 127–138. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11278021 (accessed on 12 April 2018). [CrossRef]
- Lin, H.Y.; Shih, S.C.; Chuang, C.K.; Lee, K.S.; Chen, M.R.; Lin, H.C.; Chiu, P.C.; Niu, D.M.; Lin, S.P. Assessment of hearing loss by pure-tone audiometry in patients with mucopolysaccharidoses. Mol. Genet. Metab. 2014, 111, 533–538. [Google Scholar] [CrossRef]
- Braunlin, E.A.; Harmatz, P.R.; Scarpa, M.; Furlanetto, B.; Kampmann, C.; Loehr, J.P.; Ponder, K.P.; Roberts, W.C.; Rosenfeld, H.M.; Giugliani, R. Cardiac disease in patients with mucopolysaccharidosis: Presentation, diagnosis and management. J. Inherit. Metab. Dis. 2011, 34, 1183–1197. Available online: http://www.ncbi.nlm.nih.gov/pubmed/21744090 (accessed on 12 April 2018). [CrossRef] [Green Version]
- Braunlin, E.; Wang, R. Cardiac issues in adults with the mucopolysaccharidoses: Current knowledge and emerging needs. Heart 2016, 102, 1257–1262. [Google Scholar] [CrossRef]
- Fesslová, V.; Corti, P.; Sersale, G.; Rovelli, A.; Russo, P.; Mannarino, S.; Butera, G.; Parini, R. The natural course and the impact of therapies of cardiac involvement in the mucopolysaccharidoses. Cardiol. Young 2009, 19, 170–178. Available online: http://www.journals.cambridge.org/abstract_S1047951109003576 (accessed on 4 March 2018). [CrossRef]
- De Poswar, F.O.; De Souza, C.F.M.; Giugliani, R.; Baldo, G. Aortic root dilatation in patients with mucopolysaccharidoses and the impact of enzyme replacement therapy. Heart Vessel. 2019, 34, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, J.L.; Biswas, S.; Wraith, E.; Lloyd, I.C. The ocular features of the mucopolysaccharidoses. Eye 2006, 20, 553–563. [Google Scholar] [CrossRef]
- Matheus, M.G.; Castillo, M.; Smith, J.K.; Armao, D.; Towle, D.; Muenzer, J. Brain MRI findings in patients with mucopolysaccharidosis types I and II and mild clinical presentation. Neuroradiology 2004, 46, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Parano, E.; Trifiletti, R.R.; Fiumara, A.; Pavone, P. White matter changes mimicking a leukodystrophy in a patient with Mucopolysaccharidosis: Characterization by MRI. J. Neurol. Sci. 2002, 195, 171–175. [Google Scholar] [CrossRef]
- Antunes, L.A.A.; Nogueira, A.P.B.; Castro, G.F.; Ribeiro, M.G.; De Souza, I.P.R. Dental findings and oral health status in patients with mucopolysaccharidosis: A case series. Acta Odontol. Scand. 2013, 71, 157–167. [Google Scholar] [CrossRef]
- Kubaski, F.; Osago, H.; Mason, R.; Yamaguchi, S.; Kobayashi, H.; Tsuchiya, M.; Orii, T.; Tomatsu, S. Glycosaminoglycans detection methods: Applications of mass spectrometry. Mol. Genet. Metab. 2017, 20, 67–77. Available online: http://www.ncbi.nlm.nih.gov/pubmed/27746032 (accessed on 22 October 2019). [CrossRef] [Green Version]
- Kubaski, F.; Suzuki, Y.; Orii, K.; Giugliani, R.; Church, H.J.; Mason, R.W.; Dũng, V.C.; Ngoc, C.T.; Yamaguchi, S.; Kobayashi, H.; et al. Glycosaminoglycan levels in dried blood spots of patients with mucopolysaccharidoses and mucolipidoses. Mol. Genet. Metab. 2017, 120, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Kubaski, F.; Mason, R.W.; Nakatomi, A.; Shintaku, H.; Xie, L.; Van Vlies, N.N.; Church, H.; Giugliani, R.; Kobayashi, H.; Yamaguchi, S.; et al. Newborn screening for mucopolysaccharidoses: A pilot study of measurement of glycosaminoglycans by tandem mass spectrometry. J. Inherit. Metab. Dis. 2017, 40, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, M.; Hoshina, H.; Sawamoto, K.; Kubaski, F.; Mason, R.W.; Mackenzie, W.G.; Theroux, M.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; et al. Critical review of current MPS guidelines and management. Mol. Genet. Metab. 2019, 126, 238–245. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30143438 (accessed on 3 September 2019). [CrossRef]
- Ebner, H.; Lindenschimidt, W.; Runge, H. Advantages of the combined alcian blue with periodic acid-Schiff reaction in gynecologic histopathology. Dtsch. Med. Wochenschr. 1956, 81, 1525–1529. Available online: http://www.ncbi.nlm.nih.gov/pubmed/13375227 (accessed on 22 October 2019).
- Teller, W.; Ziemann, A. Thin layer chromatography of urinary acid glycosaminoglycans as screening procedure for mucopolysaccharidoses. Horm. Metab. Res. 1969, 1, 32–35. Available online: http://www.ncbi.nlm.nih.gov/pubmed/4258873 (accessed on 25 June 2018). [CrossRef] [PubMed]
- Berman, E.R.; Vered, J.; Bach, G. A reliable spot test for mucopolysaccharidoses. Clin. Chem. 1971, 17, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Humbel, R.; Etringer, S. Colorimetric method for determination of sulfated glycosaminoglycans. Rev. Roum. Biochim. 1974, 11, 21–24. Available online: https://scholar.google.com/scholar_lookup?journal=Rev+Roum+Biochim&title=A+colorimetric+method+for+the+determination+of+sulfated+glycosaminoglycans&author=R+Humbel&author=S+Etringer&volume=11&publication_year=1974&pages=21-24& (accessed on 22 October 2019).
- Oguma, T.; Toyoda, H.; Toida, T.; Imanari, T. Analytical method for keratan sulfates by high-performance liquid chromatography/turbo-ionspray tandem mass spectrometry. Anal. Biochem. 2001, 290, 68–73. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11180938 (accessed on 7 March 2018). [CrossRef]
- Oguma, T.; Toyoda, H.; Toida, T.; Imanari, T. Analytical method of heparan sulfates using high-performance liquid chromatography turbo-ionspray ionization tandem mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 2001, 754, 153–159. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11318410 (accessed on 26 February 2018). [CrossRef]
- Oguma, T.; Toyoda, H.; Toida, T.; Imanari, T. Analytical method of chondroitin/dermatan sulfates using high performance liquid chromatography/turbo ionspray ionization mass spectrometry: Application to analyses of the tumor tissue sections on glass slides. Biomed. Chromatogr. 2001, 15, 356–362. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11507718 (accessed on 22 October 2019). [CrossRef]
- Stone, J.E. Urine analysis in the diagnosis of mucopolysaccharide disorders. Ann. Clin. Biochem. 1998, 35 Pt 2, 207–225. Available online: http://acb.sagepub.com/lookup/doi/10.1177/000456329803500204 (accessed on 7 March 2018). [CrossRef] [Green Version]
- De Jong, J.G.; Wevers, R.A.; Laarakkers, C.; Poorthuis, B.J. Dimethylmethylene blue-based spectrophotometry of glycosaminoglycans in untreated urine: A rapid screening procedure for mucopolysaccharidoses. Clin. Chem. 1989, 35, 1472–1477. Available online: http://www.ncbi.nlm.nih.gov/pubmed/2503262 (accessed on 22 October 2019). [CrossRef]
- Panin, G.; Naia, S.; Dall’Amico, R.; Chiandetti, L.; Zachello, F.; Catassi, C.; Felici, L.; Coppa, G.V. Simple spectrophotometric quantification of urinary excretion of glycosaminoglycan sulfates. Clin. Chem. 1986, 32, 2073–2076. Available online: http://www.ncbi.nlm.nih.gov/pubmed/3096595 (accessed on 22 October 2019). [CrossRef]
- Whitley, C.B.; Ridnour, M.D.; Draper, K.A.; Dutton, C.M.; Neglia, J.P. Diagnostic test for mucopolysaccharidosis. I. Direct method for quantifying excessive urinary glycosaminoglycan excretion. Clin. Chem. 1989, 35, 374–379. Available online: http://www.ncbi.nlm.nih.gov/pubmed/2493341 (accessed on 7 March 2018). [CrossRef]
- Piraud, M.; Maire, I.; Mathieu, M. Pitfalls of screening for mucopolysaccharidoses by the dimethylmethylene blue test. Clin. Chem. 1993, 39, 163–164. Available online: http://www.ncbi.nlm.nih.gov/pubmed/8419045 (accessed on 22 October 2019). [CrossRef] [PubMed]
- Kiselova, N.; Dierker, T.; Spillmann, D.; Ramström, M. An automated mass spectrometry-based screening method for analysis of sulfated glycosaminoglycans. Biochem. Biophys. Res. Commun. 2014, 450, 598–603. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0006291X14010857 (accessed on 27 October 2019). [CrossRef] [PubMed]
- Auray-Blais, C.; Bhérer, P.; Gagnon, R.; Young, S.P.; Zhang, H.H.; An, Y.; Clarke, J.T.; Millington, D.S. Efficient analysis of urinary glycosaminoglycans by LC-MS/MS in mucopolysaccharidoses type I, II and VI. Mol. Genet. Metab. 2011, 102, 49–56. Available online: http://linkinghub.elsevier.com/retrieve/pii/S1096719210003446 (accessed on 7 March 2018). [CrossRef] [PubMed]
- Oguma, T.; Tomatsu, S.; Montano, A.M.; Okazaki, O. Analytical method for the determination of disaccharides derived from keratan, heparan, and dermatan sulfates in human serum and plasma by high-performance liquid chromatography/turbo ionspray ionization tandem mass spectrometry. Anal. Biochem. 2007, 368, 79–86. Available online: http://linkinghub.elsevier.com/retrieve/pii/S0003269707003302 (accessed on 26 February 2018). [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Oguma, T.; Dung, V.C.; Oikawa, H.; De Carvalho, T.G.; Gutiérrez, M.L.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; et al. Dermatan sulfate and heparan sulfate as a biomarker for mucopolysaccharidosis I. J. Inherit. Metab. Dis. 2010, 33, 141–150. Available online: http://www.ncbi.nlm.nih.gov/pubmed/20162367 (accessed on 7 March 2018). [CrossRef]
- Osago, H.; Shibata, T.; Hara, N.; Kuwata, S.; Kono, M.; Uchio, Y.; Tsuchiya, M. Quantitative analysis of glycosaminoglycans, chondroitin/dermatan sulfate, hyaluronic acid, heparan sulfate, and keratan sulfate by liquid chromatography-electrospray ionization-tandem mass spectrometry. Anal. Biochem. 2014, 467, 62–74. Available online: http://linkinghub.elsevier.com/retrieve/pii/S0003269714003388 (accessed on 6 March 2018). [CrossRef]
- Zhang, H.; Young, S.P.; Auray-Blais, C.; Orchard, P.J.; Tolar, J.; Millington, D.S. Analysis of glycosaminoglycans in cerebrospinal fluid from patients with mucopolysaccharidoses by isotope-dilution ultra-performance liquid chromatography-tandem mass spectrometry. Clin. Chem. 2011, 57, 1005–1012. Available online: http://www.clinchem.org/cgi/doi/10.1373/clinchem.2010.161141 (accessed on 7 March 2018). [CrossRef]
- Lawrence, R.; Brown, J.R.; Al-Mafraji, K.; Lamanna, W.C.; Beitel, J.R.; Boons, G.-J.; Esko, J.D.; Crawford, B.E. Disease-specific non–reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat. Chem. Biol. 2012, 8, 197–204. Available online: http://www.ncbi.nlm.nih.gov/pubmed/22231271 (accessed on 26 February 2018). [CrossRef] [Green Version]
- Saville, J.T.; McDermott, B.K.; Fletcher, J.M.; Fuller, M. Disease and subtype specific signatures enable precise diagnosis of the mucopolysaccharidoses. Genet. Med. 2019, 21, 753–757. Available online: http://www.nature.com/articles/s41436-018-0136-z (accessed on 27 October 2019). [CrossRef]
- Kunin-Batson, A.S.; Shapiro, E.G.; Rudser, K.D.; Lavery, C.A.; Bjoraker, K.J.; Jones, S.A.; Wynn, R.F.; Vellodi, A.; Tolar, J.; Orchard, P.J.; et al. Long-term cognitive and functional outcomes in children with mucopolysaccharidosis (MPS)-IH (hurler syndrome) treated with hematopoietic cell transplantation. JIMD Rep. 2016, 29, 95–102. Available online: http://www.ncbi.nlm.nih.gov/pubmed/26825088 (accessed on 15 April 2018).
- Donati, M.A.; Pasquini, E.; Spada, M.; Polo, G.; Burlina, A. Newborn screening in mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44 (Suppl. 2), 126. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30442156 (accessed on 27 October 2019). [CrossRef] [PubMed]
- Gelb, M. Newborn screening for lysosomal storage diseases: Methodologies, screen positive rates, normalization of datasets, second-tier tests, and post-analysis tools. Int. J. Neonatal Screen. 2018, 4, 23. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30882045 (accessed on 27 October 2019). [CrossRef] [PubMed] [Green Version]
- Hopwood, J.J.; Muller, V.; Smithson, A.; Baggett, N. A fluorometric assay using 4-methylumbelliferyl α-l-iduronide for the estimation of α-l-iduronidase activity and the detection of Hurler and Scheie syndromes. Clin. Chim. Acta 1979, 92, 257–265. Available online: http://www.ncbi.nlm.nih.gov/pubmed/114339 (accessed on 27 October 2019). [CrossRef]
- Stirling, J.L.; Robinson, D.; Fensom, A.H.; Benson, P.F.; Baker, J.E. Fluorimetric assay for prenatal detection of Hurler and Scheie homozygotes or heterozygotes. Lancet 1978, 1, 147. Available online: http://www.ncbi.nlm.nih.gov/pubmed/87574 (accessed on 27 October 2019). [CrossRef]
- Pollack, M.G.; Pamula, V.K.; Srinivasan, V.; Eckhardt, A.E. Applications of electrowetting-based digital microfluidics in clinical diagnostics. Expert Rev. Mol. Diagn. 2011, 11, 393–407. [Google Scholar] [CrossRef]
- Millington, D.; Norton, S.; Singh, R.; Sista, R.; Srinivasan, V.; Pamula, V. Digital microfluidics comes of age: High-throughput screening to bedside diagnostic testing for genetic disorders in newborns. Expert Rev. Mol. Diagn. 2018, 18, 701–712. [Google Scholar] [CrossRef]
- Sista, R.S.; Wang, T.; Wu, N.; Graham, C.; Eckhardt, A.; Winger, T.; Srinivasan, V.; Bali, D.; Millington, D.S.; Pamula, V.K. Multiplex newborn screening for Pompe, Fabry, Hunter, Gaucher, and Hurler diseases using a digital microfluidic platform. Clin. Chim. Acta 2013, 424, 12–18. Available online: http://www.ncbi.nlm.nih.gov/pubmed/23660237 (accessed on 27 August 2019). [CrossRef] [Green Version]
- Kumar, A.B.; Masi, S.; Ghomashchi, F.; Chennamaneni, N.K.; Ito, M.; Scott, C.R.; Turecek, F.; Gelb, M.H.; Spacil, Z. Tandem mass spectrometry has a larger analytical range than fluorescence assays of lysosomal enzymes: Application to newborn screening and diagnosis of mucopolysaccharidoses types II, IVA, and VI. Clin. Chem. 2015, 61, 1363–1371. Available online: http://www.clinchem.org/cgi/doi/10.1373/clinchem.2015.242560 (accessed on 21 February 2018). [CrossRef]
- Blanchard, S.; Sadilek, M.; Scott, C.R.; Turecek, F.; Gelb, M.H. Tandem mass spectrometry for the direct assay of lysosomal enzymes in dried blood spots: Application to screening newborns for mucopolysaccharidosis I. Clin. Chem. 2008, 54, 2067–2070. Available online: http://www.ncbi.nlm.nih.gov/pubmed/19042989 (accessed on 12 April 2018). [CrossRef]
- Lin, S.-P.; Lin, H.-Y.; Wang, T.-J.; Chang, C.-Y.; Lin, C.-H.; Huang, S.-F.; Tsai, C.C.; Liu, H.L.; Keutzer, J.; Chuang, C.K. A pilot newborn screening program for Mucopolysaccharidosis type I in Taiwan. Orphanet J. Rare Dis. 2013, 8, 147. Available online: http://ojrd.biomedcentral.com/articles/10.1186/1750-1172-8-147 (accessed on 21 February 2018). [CrossRef] [Green Version]
- Aronovich, E.L.; Pan, D.; Whitley, C.B. Molecular genetic defect underlying appha-l-iduronidase pseudodeficiency. Am. J. Hum. Genet. 1996, 58, 75–85. [Google Scholar] [PubMed]
- Clarke, L.A.; Atherton, A.M.; Burton, B.K.; Day-Salvatore, D.L.; Kaplan, P.; Leslie, N.D.; Scott, C.R.; Stockton, D.W.; Thomas, J.A.; Muenzer, J. Mucopolysaccharidosis type I newborn screening: Best practices for diagnosis and management. J. Pediatr. 2017, 182, 363–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, L.A.; Giugliani, R.; Guffon, N.; Jones, S.A.; Keenan, H.A.; Munoz-Rojas, M.V.; Okuyama, T.; Viskochil, D.; Whitley, C.B.; Wijburg, F.A.; et al. Genotype-phenotype relationships in mucopolysaccharidosis type I (MPS I): Insights from the International MPS I Registry. Clin. Genet. 2019, 96, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusius-Facchin, A.C.; Siebert, M.; Leão, D.; Malaga, D.R.; Pasqualim, G.; Trapp, F.; Matte, U.; Giugliani, R.; Leistner-Segal, S. Phenotype-oriented NGS panels for mucopolysaccharidoses: Validation and potential use in the diagnostic flowchart. Genet. Mol. Biol. 2019, 42 (Suppl. 1), 207–214. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poletto, E.; Pasqualim, G.; Giugliani, R.; Matte, U.; Baldo, G. Worldwide distribution of common IDUA pathogenic variants. Clin. Genet. 2018, 94, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Breen, C.; Mercer, J.; Jones, S.A.; Jahic, A.; Heptinstall, L.; Tylee, K.; Newman, W.G.; Beetz, C. Maternal mosaicism for IDUA deletion clarifies recurrence risk in MPS I. Hum. Genome Var. 2016, 3, 1–3. [Google Scholar] [CrossRef]
- Yogalingam, G.; Guo, X.H.; Muller, V.J.; Brooks, D.A.; Clements, P.R.; Kakkis, E.D.; Hopwood, J.J. Identification and molecular characterization of α-l-iduronidase mutations present in mucopolysaccharidosis type I patients undergoing enzyme replacement therapy. Hum. Mutat. 2004, 24, 199–207. [Google Scholar] [CrossRef]
- Hobbs, J.R.; Hugh-Jones, K.; Barrett, A.J.; Byrom, N.; Chambers, D.; Henry, K.; James, D.C.; Lucas, C.F.; Rogers, T.R.; Benson, P.F.; et al. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 1981, 2, 709–712. Available online: http://www.ncbi.nlm.nih.gov/pubmed/6116856 (accessed on 21 April 2018). [CrossRef]
- Krivit, W.; Whitley, C.B.C.P. Lysosomal storage diseases treated by bone marrow transplantation. In Bone Marrow Transplantation: Current Controversies; Gale, R.P., Chaplin, R., Eds.; Alan Riss Inc.: New York, NY, USA, 1989; pp. 367–378. [Google Scholar]
- Hoogerbrugge, P.M.; Brouwer, O.F.; Bordigoni, P.; Ringden, O.; Kapaun, P.; Ortega, J.J.; O’Meara, A.; Cornu, G.; Souillet, G.; Frappaz, D.; et al. Allogeneic bone marrow transplantation for lysosomal storage diseases. The European Group for Bone Marrow Transplantation. Lancet 1995, 345, 1398–1402. Available online: http://www.ncbi.nlm.nih.gov/pubmed/7760610 (accessed on 21 October 2019). [CrossRef]
- Krivit, W.; Sung, J.H.; Shapiro, E.G.; Lockman, L.A. Microglia: The effector cell for reconstitution of the central nervous system following bone marrow transplantation for lysosomal and peroxisomal storage diseases. Cell Transplant. 1995, 4, 385–392. Available online: http://www.ncbi.nlm.nih.gov/pubmed/7582569 (accessed on 26 February 2018). [CrossRef] [PubMed]
- Araya, K.; Sakai, N.; Mohri, I.; Kagitani-Shimono, K.; Okinaga, T.; Hashii, Y.; Ohta, H.; Nakamichi, I.; Aozasa, K.; Taniike, M.; et al. Localized donor cells in brain of a Hunter disease patient after cord blood stem cell transplantation. Mol. Genet. Metab. 2009, 98, 255–263. [Google Scholar] [CrossRef] [PubMed]
- De Ru, M.H.; Boelens, J.J.; Das, A.M.; Jones, S.A.; Van der Lee, J.H.; Mahlaoui, N.; Mengel, E.; Offringa, M.; O’Meara, A.; Parini, R.; et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: Results of a European consensus procedure. Orphanet J. Rare Dis. 2011, 6, 55. Available online: http://www.ncbi.nlm.nih.gov/pubmed/21831279 (accessed on 21 October 2019). [CrossRef] [PubMed] [Green Version]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses: Past, present, and future. Biol. Blood Marrow Transplant. 2019, 25, e226–e246. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1083879119301375 (accessed on 3 September 2019). [CrossRef]
- Aldenhoven, M.; Van Den Broek, B.T.A.; Wynn, R.F.; O’Meara, A.; Veys, P.; Rovelli, A.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Quality of life of Hurler syndrome patients after successful hematopoietic stem cell transplantation. Blood Adv. 2017, 1, 2236–2242. [Google Scholar] [CrossRef]
- Wynn, R.F.; Wraith, J.E.; Mercer, J.; O’Meara, A.; Tylee, K.; Thornley, M.; Church, H.J.; Bigger, B.W. Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. J. Pediatr. 2009, 154, 609–611. Available online: http://linkinghub.elsevier.com/retrieve/pii/S0022347608009621 (accessed on 12 April 2018). [CrossRef]
- Boelens, J.J.; Aldenhoven, M.; Purtill, D.; Ruggeri, A.; Defor, T.; Wynn, R.; Wraith, E.; Cavazzana-Calvo, M.; Rovelli, A.; Fischer, A.; et al. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood 2013, 121, 3981–3987. Available online: http://www.ncbi.nlm.nih.gov/pubmed/23493783 (accessed on 21 October 2019). [CrossRef]
- Aldenhoven, M.; Jones, S.A.; Bonney, D.; Borrill, R.E.; Coussons, M.; Mercer, J.; Bierings, M.B.; Versluys, B.; Van Hasselt, P.M.; Wijburg, F.A.; et al. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: Results after implementation of international guidelines. Biol. Blood Marrow Transplant. 2015, 21, 1106–1109. Available online: http://linkinghub.elsevier.com/retrieve/pii/S1083879115001184 (accessed on 15 April 2018). [CrossRef] [Green Version]
- Boelens, J.J.; Van Hasselt, P.M. Neurodevelopmental outcome after hematopoietic cell transplantation in inborn errors of metabolism: Current considerations and future perspectives. Neuropediatrics 2016, 47, 285–292. Available online: http://www.thieme-connect.de/DOI/DOI?10.1055/s-0036-1584602 (accessed on 3 September 2019).
- Bartelink, I.H.; Van Reij, E.M.L.; Gerhardt, C.E.; Van Maarseveen, E.M.; De Wildt, A.; Versluys, B.; Lindemans, C.A.; Bierings, M.B.; Boelens, J.J. Fludarabine and exposure-targeted busulfan compares favorably with busulfan/cyclophosphamide-based regimens in pediatric hematopoietic cell transplantation: Maintaining efficacy with less toxicity. Biol. Blood Marrow Transplant. 2014, 20, 345–353. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1083879113005648 (accessed on 3 September 2019). [CrossRef] [Green Version]
- Wraith, J.E.; Clarke, L.A.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Rapoport, D.M.; Berger, K.I.; Swiedler, S.J.; Kakkis, E.D.; et al. Enzyme replacement therapy for mucopolysaccharidosis I: A randomized, double-blinded, placebo-controlled, multinational study of recombinant human α-l-iduronidase (laronidase). J. Pediatr. 2004, 144, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Jameson, E.; Jones, S.; Remmington, T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst. Rev. 2019, 6, CD009354. Available online: http://www.ncbi.nlm.nih.gov/pubmed/31211405 (accessed on 21 October 2019). [CrossRef] [PubMed]
- Dornelles, A.D.; De Camargo Pinto, L.L.; De Paula, A.C.; Steiner, C.E.; Lourenço, C.M.; Kim, C.A.; Horovitz, D.D.; Ribeiro, E.M.; Valadares, E.R.; Goulart, I.; et al. Enzyme replacement therapy for Mucopolysaccharidosis Type I among patients followed within the MPS Brazil Network. Genet. Mol. Biol. 2014, 37, 23–29. Available online: http://www.ncbi.nlm.nih.gov/pubmed/24688287 (accessed on 21 October 2019). [CrossRef] [PubMed] [Green Version]
- Laraway, S.; Mercer, J.; Jameson, E.; Ashworth, J.; Hensman, P.; Jones, S.A. Outcomes of long-term treatment with laronidase in patients with mucopolysaccharidosis type I. J. Pediatr. 2016, 178, 219–226. Available online: https://www.sciencedirect.com/science/article/pii/S0022347616307004 (accessed on 16 April 2018). [CrossRef] [PubMed] [Green Version]
- De Ru, M.H.; Van der Tol, L.; Van Vlies, N.; Bigger, B.W.; Hollak, C.E.M.; IJlst, L.; Kulik, W.; Van Lenthe, H.; Saif, M.A.; Wagemans, T.; et al. Plasma and urinary levels of dermatan sulfate and heparan sulfate derived disaccharides after long-term enzyme replacement therapy (ERT) in MPS I: Correlation with the timing of ERT and with total urinary excretion of glycosaminoglycans. J. Inherit. Metab. Dis. 2013, 36, 247–255. Available online: http://link.springer.com/10.1007/s10545-012-9538-2 (accessed on 26 February 2018). [CrossRef] [PubMed] [Green Version]
- Clarke, L.A.; Wraith, J.E.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Rapoport, D.M.; Berger, K.I.; Sidman, M.; Kakkis, E.D.; et al. Long-term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics 2009, 123, 229–240. Available online: http://www.ncbi.nlm.nih.gov/pubmed/19117887 (accessed on 12 April 2018). [CrossRef]
- Pal, A.R.; Langereis, E.J.; Saif, M.A.; Mercer, J.; Church, H.J.; Tylee, K.L.; Wynn, R.F.; Wijburg, F.A.; Jones, S.A.; Bruce, I.A.; et al. Sleep disordered breathing in mucopolysaccharidosis I: A multivariate analysis of patient, therapeutic and metabolic correlators modifying long term clinical outcome. Orphanet J. Rare Dis. 2015, 10, 42. Available online: http://www.ojrd.com/content/10/1/42 (accessed on 26 February 2018). [CrossRef] [Green Version]
- Al-Sannaa, N.A.; Bay, L.; Barbouth, D.S.; Benhayoun, Y.; Goizet, C.; Guelbert, N.; Jones, S.A.; Kyosen, S.O.; Martins, A.M.; Phornphutkul, C. Early treatment with laronidase improves clinical outcomes in patients with attenuated MPS I: A retrospective case series analysis of nine sibships. Orphanet J. Rare Dis. 2015, 10, 131. Available online: http://www.ojrd.com/content/10/1/131 (accessed on 30 January 2018). [CrossRef] [Green Version]
- Sifuentes, M.; Doroshow, R.; Hoft, R.; Mason, G.; Walot, I.; Diament, M.; Okazaki, S.; Huff, K.; Cox, G.F.; Swiedler, S.J.; et al. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol. Genet. Metab. 2007, 90, 171–180. Available online: http://www.ncbi.nlm.nih.gov/pubmed/17011223 (accessed on 21 October 2019). [CrossRef]
- Wang, R.Y.; Cambray-Forker, E.J.; Ohanian, K.; Karlin, D.S.; Covault, K.K.; Schwartz, P.H.; Abdenur, J.E. Treatment reduces or stabilizes brain imaging abnormalities in patients with MPS I and II. Mol. Genet. Metab. 2009, 98, 406–411. Available online: http://www.ncbi.nlm.nih.gov/pubmed/19748810 (accessed on 26 February 2018). [CrossRef]
- Boado, R.J.; Zhang, Y.; Zhang, Y.; Xia, C.F.; Wang, Y.; Pardridge, W.M. Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood-brain barrier. Biotechnol. Bioeng. 2008, 99, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Pardridge, W.M. Brain and organ uptake in the Rhesus monkey in vivo of recombinant iduronidase compared to an insulin receptor antibody-iduronidase fusion protein. Mol. Pharm. 2017, 14, 1271–1277. [Google Scholar] [CrossRef]
- Boado, R.J.; Hui, E.K.W.; Lu, J.Z.; Zhou, Q.H.; Pardridge, W.M. Reversal of lysosomal storage in brain of adult MPS-I mice with intravenous Trojan horse-iduronidase fusion protein. Mol. Pharm. 2011, 8, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Giugliani, L.; De Oliveira Poswar, F.; Donis, K.C.; Corte, A.D.; Schmidt, M.; Boado, R.J.; Nestrasil, I.; Nguyen, C.; Chen, S.; et al. Neurocognitive and somatic stabilization in pediatric patients with severe Mucopolysaccharidosis Type I after 52 weeks of intravenous brain-penetrating insulin receptor antibody-iduronidase fusion protein (valanafusp alpha): An open label phase 1–2 trial. Orphanet J. Rare Dis. 2018, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, T.; Eto, Y.; Sakai, N.; Minami, K.; Yamamoto, T.; Sonoda, H.; Yamaoka, M.; Tachibana, K.; Hirato, T.; Sato, Y. Iduronate-2-sulfatase with anti-human transferrin receptor antibody for neuropathic mucopolysaccharidosis II: A phase 1/2 trial. Mol. Ther. 2019, 27, 456–464. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30595526 (accessed on 6 February 2019). [CrossRef] [PubMed] [Green Version]
- Munoz-Rojas, M.V.; Vieira, T.; Costa, R.; Fagondes, S.; John, A.; Jardim, L.B.; Vedolin, L.M.; Raymundo, M.; Dickson, P.I.; Kakkis, E.; et al. Intrathecal enzyme replacement therapy in a patient with mucopolysaccharidosis type I and symptomatic spinal cord compression. Am. J. Med. Genet. Part A 2008, 146, 2538–2544. [Google Scholar] [CrossRef]
- Dickson, P.I.; Kaitila, I.; Harmatz, P.; Mlikotic, A.; Chen, A.H.; Victoroff, A.; Passage, M.B.; Madden, J.; Le, S.Q.; Naylor, D.E.; et al. Safety of laronidase delivered into the spinal canal for treatment of cervical stenosis in mucopolysaccharidosis I. Mol. Genet. Metab. 2015, 116, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Nestrasil, I.; Shapiro, E.; Svatkova, A.; Dickson, P.; Chen, A.; Wakumoto, A.; Ahmed, A.; Stehel, E.; McNeil, S.; Gravance, C.; et al. Intrathecal enzyme replacement therapy reverses cognitive decline in mucopolysaccharidosis type I. Am. J. Med. Genet. Part A 2017, 173, 780–783. [Google Scholar] [CrossRef] [Green Version]
- Traas, A.M.; Wang, P.; Ma, X.; Tittiger, M.; Schaller, L.; O’donnell, P.; Sleeper, M.M.; Vite, C.; Herati, R.; Aguirre, G.D.; et al. Correction of clinical manifestations of canine mucopolysaccharidosis I with neonatal retroviral vector gene therapy. Mol. Ther. 2007, 15, 1423–1431. Available online: http://www.ncbi.nlm.nih.gov/pubmed/17519893 (accessed on 21 October 2019). [CrossRef]
- Hinderer, C.; Bell, P.; Louboutin, J.P.; Zhu, Y.; Yu, H.; Lin, G.; Choa, R.; Gurda, B.L.; Bagel, J.; O’Donnell, P.; et al. Neonatal systemic AAV induces tolerance to CNS gene therapy in MPS I dogs and nonhuman primates. Mol. Ther. 2015, 23, 1298–1307. [Google Scholar] [CrossRef] [Green Version]
- Ou, L.; DeKelver, R.C.; Rohde, M.; Tom, S.; Radeke, R.; St Martin, S.J.; Santiago, Y.; Sproul, S.; Przybilla, M.J.; Koniar, B.L.; et al. ZFN-mediated in vivo genome editing corrects murine hurler syndrome. Mol. Ther. 2019, 27, 178–187. [Google Scholar] [CrossRef]
- Sheridan, C. Sangamo’s landmark genome editing trial gets mixed reception. Nat. Biotechnol. 2018, 36, 907–908. [Google Scholar] [CrossRef]
- Gomez-Ospina, N.; Scharenberg, S.G.; Mostrel, N.; Bak, R.O.; Mantri, S.; Quadros, R.M.; Gurumurthy, C.B.; Lee, C.; Bao, G.; Suarez, C.J.; et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunn, G.; Dai, Y.; Du, M.; Belakhov, V.; Kandasamy, J.; Schoeb, T.R.; Baasov, T.; Bedwell, D.M.; Keeling, K.M. Long-term nonsense suppression therapy moderates MPS I-H disease progression. Mol. Genet. Metab. 2014, 111, 374–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamei, M.; Kasperski, K.; Fuller, M.; Parkinson-Lawrence, E.J.; Karageorgos, L.; Belakhov, V.; Baasov, T.; Hopwood, J.J.; Brooks, D.A. Aminoglycoside-induced premature stop codon read-through of mucopolysaccharidosis type I patient Q70X and W402X mutations in cultured cells. JIMD Rep. 2014, 13, 139–147. Available online: http://www.ncbi.nlm.nih.gov/pubmed/24193436 (accessed on 6 November 2019).
- Smith, L.J.; Martin, J.T.; O’Donnell, P.; Wang, P.; Elliott, D.M.; Haskins, M.E.; Ponder, K.P. Effect of neonatal gene therapy on lumbar spine disease in mucopolysaccharidosis VII dogs. Mol. Genet. Metab. 2012, 107, 145–152. Available online: http://linkinghub.elsevier.com/retrieve/pii/S109671921200114X (accessed on 20 February 2018). [CrossRef] [PubMed] [Green Version]
- Polgreen, L.E.; Kunin-Batson, A.; Rudser, K.; Vehe, R.K.; Utz, J.J.; Whitley, C.B.; Dickson, P. Pilot study of the safety and effect of adalimumab on pain, physical function, and musculoskeletal disease in mucopolysaccharidosis types I and II. Mol. Genet. Metab. Rep. 2017, 10, 75–80. Available online: https://linkinghub.elsevier.com/retrieve/pii/S2214426916300921 (accessed on 3 September 2017). [CrossRef] [PubMed]
- Hennermann, J.B.; Gökce, S.; Solyom, A.; Mengel, E.; Schuchman, E.H.; Simonaro, C.M. Treatment with pentosan polysulphate in patients with MPS I: Results from an open label, randomized, monocentric phase II study. J. Inherit. Metab. Dis. 2016, 39, 831–837. Available online: http://link.springer.com/10.1007/s10545-016-9974-5 (accessed on 20 February 2018). [CrossRef]
- Matern, D.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; Rinaldo, P.; Tortorelli, S. Newborn screening for lysosomal storage disorders. Semin. Perinatol. 2015, 39, 206–216. Available online: http://www.ncbi.nlm.nih.gov/pubmed/25891428 (accessed on 21 February 2018). [CrossRef]
- Eisengart, J.B.; Rudser, K.D.; Xue, Y.; Orchard, P.; Miller, W.; Lund, T.; Van der Ploeg, A.; Mercer, J.; Jones, S.; Mengel, K.E.; et al. Long-term outcomes of systemic therapies for Hurler syndrome: An international multicenter comparison. Genet. Med. 2018, 20, 1423–1429. [Google Scholar] [CrossRef] [Green Version]
- Parini, R.; Broomfield, A.; Cleary, M.A.; De Meirleir, L.; Di Rocco, M.; Fathalla, W.M.; Guffon, N.; Lampe, C.; Lund, A.M.; Scarpa, M.; et al. International working group identifies need for newborn screening for mucopolysaccharidosis type I but states that existing hurdles must be overcome. Acta Paediatr. 2018, 107, 2059–2065. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30242902 (accessed on 28 October 2019). [CrossRef] [PubMed]
- Chan, M.-J.; Liao, H.-C.; Gelb, M.H.; Chuang, C.-K.; Liu, M.-Y.; Chen, H.-J.; Kao, S.M.; Lin, H.Y.; Huang, Y.H.; Kumar, A.B.; et al. Taiwan national newborn screening program by tandem mass spectrometry for mucopolysaccharidoses types I, II, and VI. J. Pediatr. 2019, 205, 176–182. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30409495 (accessed on 28 October 2019). [CrossRef] [PubMed]
- Burlina, A.B.; Polo, G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro, G.; Salviati, L.; Burlina, A.P. Implementation of second-tier tests in newborn screening for lysosomal disorders in north eastern Italy. Int. J. Neonatal Screen. 2019, 5, 24. Available online: https://www.mdpi.com/2409-515X/5/2/24 (accessed on 19 August 2019). [CrossRef] [Green Version]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. Available online: http://www.ncbi.nlm.nih.gov/pubmed/29143201 (accessed on 26 June 2018). [CrossRef] [PubMed]
- Metz, T.F.; Mechtler, T.P.; Orsini, J.J.; Martin, M.; Shushan, B.; Herman, J.L.; Ratschmann, R.; Item, C.B.; Streubel, B.; Herkner, K.R.; et al. Simplified newborn screening protocol for lysosomal storage disorders. Clin. Chem. 2011, 57, 1286–1294. Available online: http://www.ncbi.nlm.nih.gov/pubmed/21771947 (accessed on 28 October 2019). [CrossRef] [Green Version]
- Bravo, H.; Neto, E.C.; Schulte, J.; Pereira, J.; Filho, C.S.; Bittencourt, F.; Sebastião, F.; Bender, F.; De Magalhães, A.P.S.; Guidobono, R.; et al. Investigation of newborns with abnormal results in a newborn screening program for four lysosomal storage diseases in Brazil. Mol. Genet. Metab. Rep. 2017, 12, 92–97. Available online: http://www.ncbi.nlm.nih.gov/pubmed/28721335 (accessed on 6 November 2019). [CrossRef]
- Eyskens, F.; Devos, S. Newborn screening for lysosomal storage disorders in Belgium. J. Inborn Errors Metab. Screen. 2017, 5, 232640981774423. [Google Scholar] [CrossRef]
- Navarrete-Martínez, J.I.; Limón-Rojas, A.E.; De Gaytán-García, M.J.; Reyna-Figueroa, J.; Wakida-Kusunoki, G.; Del Delgado-Calvillo, M.R.; Cantú-Reyna, C.; Cruz-Camino, H.; Cervantes-Barragán, D.E. Newborn screening for six lysosomal storage disorders in a cohort of Mexican patients: Three-year findings from a screening program in a closed Mexican health system. Mol. Genet. Metab. 2017, 121, 16–21. Available online: http://www.ncbi.nlm.nih.gov/pubmed/28302345 (accessed on 12 April 2018).
- Scott, C.R.; Elliott, S.; Buroker, N.; Thomas, L.I.; Keutzer, J.; Glass, M.; Gelb, M.H.; Turecek, F. Identification of infants at risk for developing fabry, pompe, or mucopolysaccharidosis-I from newborn blood spots by tandem mass spectrometry. J. Pediatr. 2013, 163, 498–503. Available online: http://www.ncbi.nlm.nih.gov/pubmed/23465405 (accessed on 12 April 2018). [CrossRef] [Green Version]
- Schielen, P.C.J.I.; Kemper, E.A.; Gelb, M.H. Newborn screening for lysosomal storage diseases: A concise review of the literature on screening methods, therapeutic possibilities and regional programs. Int. J. Neonatal Screen. 2017, 3, 6. Available online: http://www.ncbi.nlm.nih.gov/pubmed/28730181 (accessed on 26 June 2018). [CrossRef]
- Taylor, J.L.; Lee, S. Lessons learned from newborn screening in pilot studies. N. C. Med. J. 2019, 80, 54–58. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30622208 (accessed on 28 October 2019). [CrossRef] [PubMed]
- Vijay, S.; Ed Wraith, J. Clinical presentation and follow-up of patients with the attenuated phenotype of mucopolysaccharidosis type I. Acta Paediatr. 2007, 94, 872–877. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant a | Combined Frequency (%) |
|---|---|
| p.Trp402 * | 31.5 |
| p.Pro533Arg | 25.8 |
| p.Gln70 * | 21.5 |
| c.56_47del12 | 6.3 |
| p.Arg89Gln | 6.3 |
| p.Arg628 * | 5.5 |
| p.Gln380Arg | 5.1 |
| p.Ala327Pro | 4.9 |
| p.Glu178Lys | 3.4 |
| Therapy | Category | Delivery | Current Status of Development | ClinicalTrials.Gov Identifier |
|---|---|---|---|---|
| Laronidase (Aldurazyme®) | Enzyme replacement therapy | I.V. infusion | Approved | NA |
| Stem cell transplantation | Stem cell transplantation | I.V. infusion | Approved | NA |
| Intrathecal Laronidase | Intrathecal Enzyme replacement therapy | Intrathecal administration | Phase I | NCT00638547, NCT00852358 |
| SB-318 | Genome editing | I.V. infusion | Phase I/II | NCT02702115 |
| RGX-111 | Gene therapy | Intracisternal injection | Phase I | NCT03580083 |
| AGT-181 | Enzyme replacement therapy with fusion protein | I.V. infusion | Phase I/II | NCT02597114, NCT03071341, NCT02371226, NCT03053089 |
| Genetically modified autologous hematopoietic stem cells | Ex vivo gene therapy | I.V. infusion | Phase I/II | NCT03488394 |
| Genotype | Severe Phenotype (n = 380) | Attenuated Phenotype (n = 158) |
|---|---|---|
| c.1403-1G> T;p.[Gln400*] | 2 (0.5%) | 0 |
| c.1524+1G>T;p.[Trp402*] | 2 (0.5%) | 0 |
| c.1650+5G>A;p.[Trp402*] | 3 (0.8%) | 0 |
| c.1727+5G>C;p.[Asn348Lys] | 0 | 2 (1.3%) |
| c.386-2A>G;c.386-2A>G | 2 (0.5%) | 0 |
| c.386-2A>G;p.[Trp402*] | 2 (0.5%) | 0 |
| p.[Ala327Pro];[Ala327Pro] | 3 (0.8%) | 2 (1.3%) |
| p.[Ala327Pro];[Arg383His] | 0 | 1 (0.6%) |
| p.[Ala327Pro];[Arg89Trp] | 0 | 1 (0.6%) |
| p.[Ala327Pro];[Gln380Arg] | 0 | 1 (0.6%) |
| p.[Ala327Pro];[Gln70*] | 5 (1.3%) | 0 |
| p.[Ala327Pro];[Ser423Arg] | 1 (0.2%) | 0 |
| p.[Ala327Pro];[Thr374Asn] | 0 | 1 (0.6%) |
| p.[Ala327Pro];c.1190-1G>C | 1 (0.2%) | 0 |
| p.[Ala327Pro];[Ala327Pro] | 0 | 2 (1.3%) |
| p.[Ala327Pro];[Trp402*] | 14 (3.7%) | 0 |
| p.[Ala36Glu];[Gln70*] | 0 | 2 (1.3%) |
| p.[Ala75Thr];[Gln70*] | 2 (0.5%) | 0 |
| p.[Ala75Thr];[Trp402*] | 2 (0.5%) | 0 |
| p.[Arg383His]; c.386-2A>G | 0 | 3 (1.9%) |
| p.[Arg383His];[Gln70*] | 0 | 2 (1.3%) |
| p.[Arg383His];[Trp402*] | 0 | 2 (1.3%) |
| p.[Arg619*];[Trp402*] | 5 (1.3%) | 0 |
| p.[Arg628*];[Arg628*] | 6 (1.6%) | 0 |
| p.[Arg89Gln];[Trp402*] | 0 | 4 (2.5%) |
| p.[Arg89Trp];[Trp402*] | 0 | 3 (1.9%) |
| p.[Asn110Asp];[Gln70*] | 2 (0.5%) | 0 |
| p.[Asn348Lys];[Trp402*] | 0 | 2 (1.3%) |
| p.[Gln380*];[Arg654*] | 0 | 2 (1.3%) |
| p.[Gln380Arg];[Gln380Arg] | 0 | 4 (2.5%) |
| p.[Gln380Arg];[Thr388Arg] | 0 | 2 (1.3%) |
| p.[Gln380Arg];[Trp402*] | 0 | 2 (1.3%) |
| p.[Gln70*];[Gln70*] | 24 (6.3%) | 0 |
| p.[Gly265Arg];[Trp402*] | 0 | 2 (1.3%) |
| p.[Gly51Asp];[Gly51Asp] | 2 (0.5%) | 0 |
| p.[Leu18Pro];[Thr388Lys] | 2 (0.5%) | 0 |
| p.[Leu218P];[Gln70*] | 5 (1.3%) | 0 |
| p.[Leu218P];[Leu218P] | 2 (0.5%) | 0 |
| p.[Leu218P];[Trp402*] | 3 (0.8%) | 0 |
| p.[Leu238Gln];[Gln70*] | 0 | 2 (1.3%) |
| p.[Leu238Gln];[Trp402*] | 0 | 6 (3.8%) |
| p.[Leu490Pro];[Leu490Pro] | 0 | 21 (13.3%) |
| p.[Leu535Phe];[Trp402*] | 0 | 2 (1.3%) |
| p.[Lys153*];[Trp402*] | 2 (0.5%) | 0 |
| p.[Pro21fs];[Trp402*] | 3 (0.8%) | 0 |
| p.[Pro496Arg];[Gln70*] | 4 (1.1%) | 0 |
| p.[Pro533Arg];[Arg363Leu] | 2 (0.5%) | 0 |
| p.[Pro533Arg];[Arg621*] | 0 | 1 (0.6%) |
| p.[Pro533Arg];[Arg89Gln] | 0 | 1 (0.6%) |
| p.[Pro533Arg];[Asp184Val] | 0 | 1 (0.6%) |
| p.[Pro533Arg];[Asp301Glu] | 2 (0.5%) | 0 |
| p.[Pro533Arg];[Gln70*] | 0 | 2 (1.3%) |
| p.[Pro533Arg];[Gly197Asp] | 0 | 1 (0.6%) |
| p.[Pro533Arg];[His425Profs*84] | 1 (0.2%) | 0 |
| p.[Pro533Arg];[IleI259Asn] | 0 | 1 (0.6%) |
| p.[Pro533Arg];[Lys153*] | 0 | 1 (0.6%) |
| p.[Pro533Arg];[Pro533Arg] | 7 (1.8%) | 17 (10.8%) |
| p.[Pro533Arg];[Ser633Leu] | 0 | 1 (0.6%) |
| p.[Pro533Arg];[Trp402*] | 5 (1.3%) | 3 (1.9%) |
| p.[Pro533Arg];[Trp402*] | 0 | 3 (1.9%) |
| p.[Ser16del];[Glu178Lys] | 0 | 2 (1.3%) |
| p.[Ser16del];[Trp402*] | 6 (1.6%) | 0 |
| p.[Ser633Leu];[Trp402*] | 0 | 3 (1.9%) |
| p.[Ser633Leu];[S16_A19del] | 0 | 3 (1.9%) |
| p.[Thr388Arg];[Trp402*] | 5 (1.3%) | 0 |
| p.[Trp402*];[Gln70*] | 61 (16.1%) | 0 |
| p.[Trp402*];[Trp402*] | 109 (28.7%) | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubaski, F.; de Oliveira Poswar, F.; Michelin-Tirelli, K.; Matte, U.d.S.; Horovitz, D.D.; Barth, A.L.; Baldo, G.; Vairo, F.; Giugliani, R. Mucopolysaccharidosis Type I. Diagnostics 2020, 10, 161. https://doi.org/10.3390/diagnostics10030161
Kubaski F, de Oliveira Poswar F, Michelin-Tirelli K, Matte UdS, Horovitz DD, Barth AL, Baldo G, Vairo F, Giugliani R. Mucopolysaccharidosis Type I. Diagnostics. 2020; 10(3):161. https://doi.org/10.3390/diagnostics10030161
Chicago/Turabian StyleKubaski, Francyne, Fabiano de Oliveira Poswar, Kristiane Michelin-Tirelli, Ursula da Silveira Matte, Dafne D. Horovitz, Anneliese Lopes Barth, Guilherme Baldo, Filippo Vairo, and Roberto Giugliani. 2020. "Mucopolysaccharidosis Type I" Diagnostics 10, no. 3: 161. https://doi.org/10.3390/diagnostics10030161
APA StyleKubaski, F., de Oliveira Poswar, F., Michelin-Tirelli, K., Matte, U. d. S., Horovitz, D. D., Barth, A. L., Baldo, G., Vairo, F., & Giugliani, R. (2020). Mucopolysaccharidosis Type I. Diagnostics, 10(3), 161. https://doi.org/10.3390/diagnostics10030161