Retroperitoneal Sarcomas: An Update on the Diagnostic Pathology Approach



Abstract
:1. Introduction
2. Diagnostic Pathology Approach to Retroperitoneal Tumors
2.1. Anatomy of the Retroperitoneum
2.2. Clinical and Imaging Considerations
2.3. Retroperitoneal Tumor Specimen Handling
2.4. Histologic Evaluation
2.5. Immunohistochemistry
2.6. Molecular Testing
3. Relatively Common Retroperitoneal Sarcomas
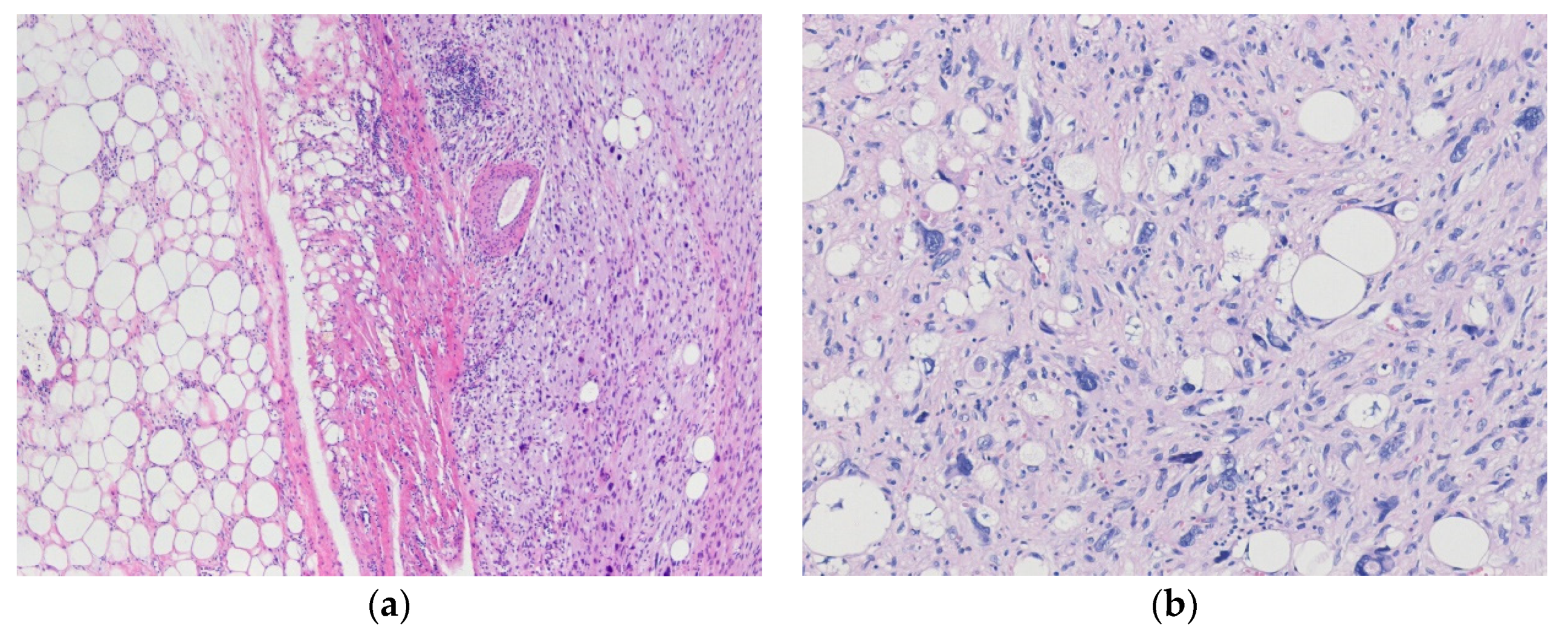
3.1. Liposarcoma
3.1.1. Well-Differentiated Liposarcoma
3.1.2. Dedifferentiated Liposarcoma
3.1.3. Pleomorphic Liposarcoma
3.1.4. Myxoid Liposarcoma
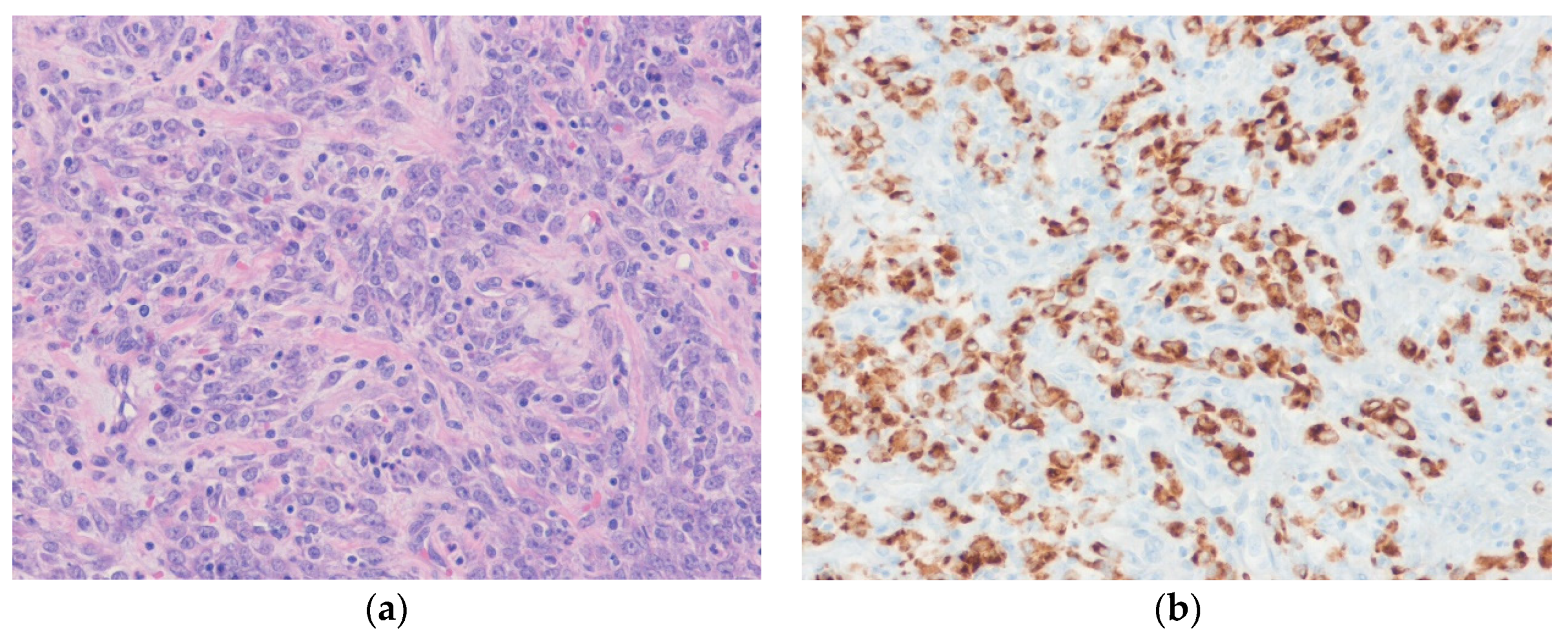
3.2. Leiomyosarcoma
4. Rare Retroperitoneal Sarcomas
4.1. Solitary Fibrous Tumor
4.2. Inflammatory Myofibroblastic Tumor
4.3. Rhabdomyosarcoma
4.4. Malignant Peripheral Nerve Sheath Tumor
4.5. Extraskeletal Osteosarcoma
4.6. Synovial Sarcoma
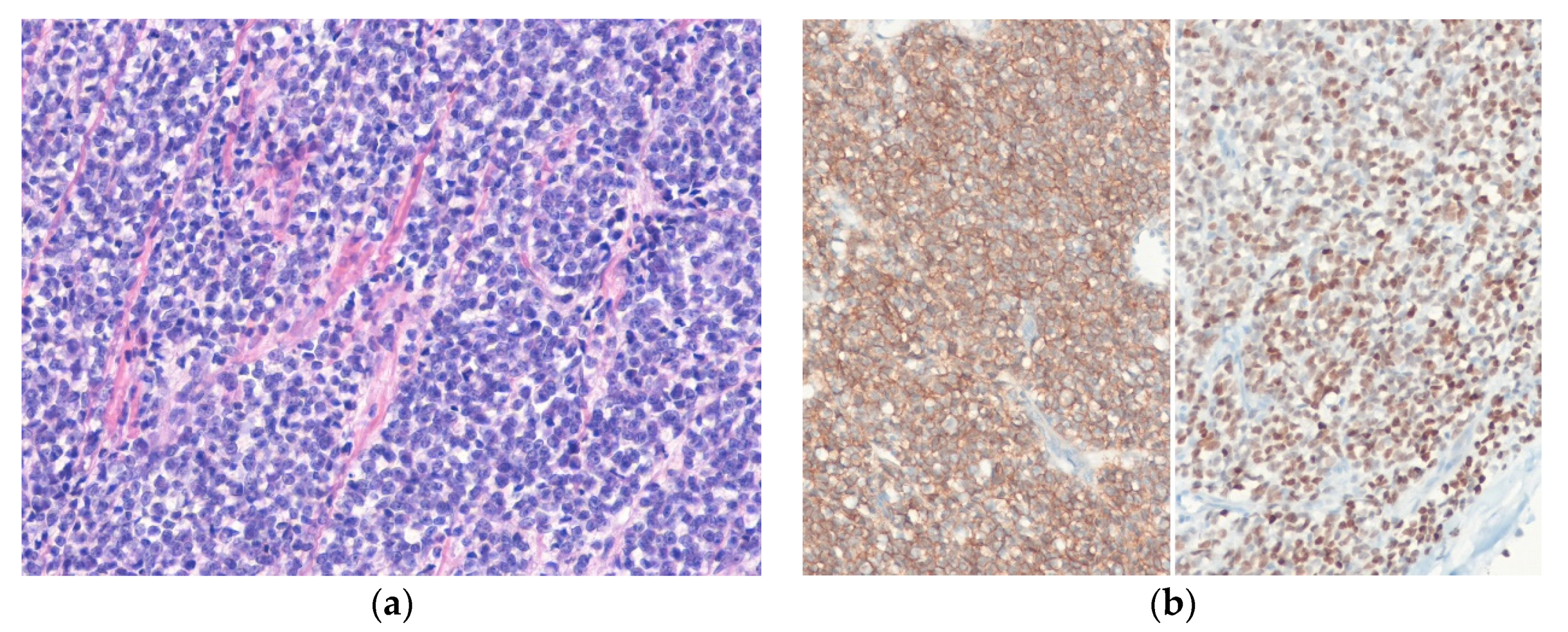
4.7. Desmoplastic Small Round Cell Tumor
4.8. PEComa
4.9. Undifferentiated Pleomorphic Sarcoma
4.10. Extraskeletal Ewing Sarcoma
5. Miscellaneous Retroperitoneal Mesenchymal Tumors
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALK | anaplastic lymphoma kinase |
| ALT | atypical lipomatous tumor |
| CDK4 | cyclin-dependent kinase 4 |
| CT | computed tomography |
| DDLPS | dedifferentiated liposarcoma |
| DOG1 | discovered on GIST 1 |
| DSRCT | desmoplastic small round cell tumor |
| EMA | epithelial membrane antigen |
| EOS | extraskeletal osteosarcoma |
| ERG | ETS-related gene |
| ES | Ewing sarcoma |
| ETS | erythroblast transformation-specific |
| FET | female expressed transcript |
| FISH | fluorescence in situ hybridization |
| FNCLCC | Fédération nationale des centres de lutte contre le cancer |
| H3K27me3 | histone 3K37 trimethylation |
| H&E stain | hematoxylin and eosin stain |
| HMB-45 | human melanoma black-45 |
| HPF | high-power field |
| IMT | inflammatory myofibroblastic tumor |
| INI1 | integrase interactor 1 |
| MDM2 | mouse double minute 2 homolog |
| MLPS | myxoid liposarcoma |
| MPNST | malignant peripheral nerve sheath tumor |
| MRI | magnetic resonance imaging |
| MYOD1 | myogenic differentiation 1 |
| NF1 | neurofibromatosis type 1 |
| NKX2.2 | NKX2 homeobox 2 |
| NSE | neuron specific enolase |
| PAX8 | paired box 8 |
| PEComa | perivascular epithelioid cell tumor |
| PLPS | pleomorphic liposarcoma |
| RMS | rhabdomyosarcoma |
| SFT | solitary fibrous tumor |
| SMA | smooth muscle actin |
| TFE3 | transcription factor E3 |
| TLE1 | transducin-like enhancer of split 1 |
| TNM | tumor, lymph node, metastasis |
| UPS | undifferentiated pleomorphic sarcoma |
| WDLPS | well-differentiated liposarcoma |
| WHO | World Health Organization |
| WT1 | Wilms’ tumor 1 |
References
- Fletcher, C.D.M.; Baldini, E.H.; Blay, J.Y.; Gronchi, A.; Lazar, A.J.; Messiou, C.; Pollock, R.E.; Singer, S. Soft tissue tumors: Introduction. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 1–12. [Google Scholar]
- Hornick, J.L. Introduction: Tumor classification and immunohistochemistry. In Practical Soft Tissue Pathology: A Diagnostic Approach, 2nd ed.; Hornick, J.L., Ed.; Elsevier: Philadelphia, PA, USA, 2019; pp. 1–6. [Google Scholar]
- The WHO Classification of Tumours Editorial Board. WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; IARC Press: Lyon, France, 2020. [Google Scholar]
- Van Roggen, J.F.; Hogendoorn, P.C. Soft tissue tumours of the retroperitoneum. Sarcoma 2000, 4, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Rosai, J. Rosai and Ackerman’s Surgical Pathology, 10th ed.; Elsevier: Philadelphia, PA, USA, 2011; pp. 2251–2258. [Google Scholar]
- Pollock, R.E.; Maki, R.G.; Baldini, E.H.; Hornick, J.L.; Keedy, V.L.; Lazar, A.J.; Madewell, J.E.; Raut, C.P.; Tedder, P.S.; Yoon, S.S. Soft tissue sarcoma of the retroperitoneum. In AJCC Caner Staging Manual, 8th ed.; Amin, M.B., Ed.; American Joint Committee on Cancer, Springer: Chicago, IL, USA, 2017; pp. 531–537. [Google Scholar]
- Cormier, J.N.; Pollock, R.E. Soft tissue sarcomas. CA. Cancer J. Clin. 2004, 54, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Turnage, R.H.; Mizell, J.; Badgwell, B. Abdominal wall, umbilicus, peritoneum, mesenteries, omentum, and retroperitoneum. In Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice, 20th ed.; Townsend, C.M., Jr., Daniel Beauchamp, R., Mark Evers, B., Mattox, K.L., Eds.; Elsevier: Philadelphia, PA, USA, 2017; pp. 1066–1089. [Google Scholar]
- Mito, J.K.; Mitra, D.; Doyle, L.A. Radiation-associated sarcomas: An update on clinical, histologic, and molecular features. Surg. Pathol. Clin. 2019, 12, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Widemann, B.C. Current status of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Curr. Oncol. Rep. 2009, 11, 322–328. [Google Scholar] [CrossRef]
- Levy, A.D.; Manning, M.A.; Al-Refaie, W.B.; Miettinen, M.M. Soft-tissue sarcomas of the abdomen and pelvis: Radiologic-pathologic features, part 1—Common sarcomas: From the radiologic pathology archives. Radiographics 2017, 37, 462–483. [Google Scholar] [CrossRef] [Green Version]
- Rajiah, P.; Sinha, R.; Cuevas, C.; Dubinsky, T.J.; Bush, W.H., Jr.; Kolokythas, O. Imaging of uncommon retroperitoneal masses. Radiographics 2011, 31, 949–976. [Google Scholar] [CrossRef]
- Thway, K.; Jordan, S.; Fisher, C.; Nicholson, A.G. Updates in the approach to intrathoracic sarcomas. Histopathology 2015, 67, 755–770. [Google Scholar] [CrossRef]
- Westra, W.H.; Hruban, R.H.; Phelps, T.H.; Isacson, C. Surgical Pathology Dissection: An Illustrated Guide, 2nd ed.; Springer: New York, NY, USA, 2003; pp. 120–123. [Google Scholar]
- Hornick, J.L. Subclassification of pleomorphic sarcomas: How and why should we care? Ann. Diagn. Pathol. 2018, 37, 118–124. [Google Scholar] [CrossRef]
- Goldblum, J.R.; Folpe, A.L.; Weiss, S.W. Approach to the diagnosis of soft tissue tumors. In Enzinger and Weiss’s Soft Tissue Tumors, 7th ed.; Elsevier: Philadelphia, PA, USA, 2020; pp. 121–128. [Google Scholar]
- Samaratunga, H.; Delahunt, B.; Srigley, J.R.; Berney, D.M.; Cheng, L.; Evans, A.; Furusato, B.; Leite, K.R.M.; MacLennan, G.T.; Martignoni, G.; et al. Granular necrosis: A distinctive form of cell death in malignant tumours. Pathology 2020. [Google Scholar] [CrossRef]
- Trojani, M.; Contesso, G.; Coindre, J.M.; Rouesse, J.; Bui, N.B.; de Mascarel, A.; Goussot, J.F.; David, M.; Bonichon, F.; Lagarde, C. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of histopathological grading system. Int. J. Cancer 1984, 33, 37–42. [Google Scholar] [CrossRef]
- Hornick, J.L. Novel uses of immunohistochemistry in the diagnosis and classification of soft tissue tumors. Mod. Pathol. 2014, 27 (Suppl. 1), S47–S63. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, I.M.; Fletcher, C.D.M. Recent advances in the diagnosis of soft tissue tumours. Pathology 2018, 50, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Borden, E.C.; Baker, L.H.; Bell, R.S.; Bramwell, V.; Demetri, G.D.; Eisenberg, B.L.; Fletcher, C.D.; Fletcher, J.A.; Ladanyi, M.; Meltzer, P.; et al. Soft tissue sarcomas of adults: State of the translational science. Clin. Cancer Res. 2003, 9, 1941–1956. [Google Scholar]
- Lahat, G.; Lazar, A.; Lev, D. Sarcoma epidemiology and etiology: Potential environmental and genetic factors. Surg. Clin. North. Am. 2008, 88, 451–481. [Google Scholar] [CrossRef] [PubMed]
- Bridge, J.A. The role of cytogenetics and molecular diagnostics in the diagnosis of soft-tissue tumors. Mod. Pathol. 2014, 27 (Suppl. 1), S80–S97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shern, J.F.; Yohe, M.E.; Khan, J. Pediatric rhabdomyosarcoma. Crit. Rev. Oncog. 2015, 20, 227–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, I.M.; Dong, F.; Garcia, E.P.; Fletcher, C.D.M.; Jo, V.Y. Recurrent SMARCB1 inactivation in epithelioid malignant peripheral nerve sheath tumors. Am. J. Surg. Pathol. 2019, 43, 835–843. [Google Scholar] [CrossRef]
- Sawyer, J.R.; Tryka, A.F.; Lewis, J.M. A novel reciprocal chromosome translocation t(11;22)(p13;q12) in an intraabdominal desmoplastic small round-cell tumor. Am. J. Surg. Pathol. 1992, 16, 411–416. [Google Scholar] [CrossRef]
- Pan, C.C.; Chung, M.Y.; Ng, K.F.; Liu, C.Y.; Wang, J.S.; Chai, C.Y.; Huang, S.H.; Chen, P.C.; Ho, D.M. Constant allelic alteration on chromosome 16p (TSC2 gene) in perivascular epithelioid cell tumour (PEComa): Genetic evidence for the relationship of PEComa with angiomyolipoma. J. Pathol. 2008, 214, 387–393. [Google Scholar] [CrossRef]
- Rao, Q.; Shen, Q.; Xia, Q.Y.; Wang, Z.Y.; Liu, B.; Shi, S.S.; Shi, Q.L.; Yin, H.L.; Wu, B.; Ye, S.B.; et al. PSF/SFPQ is a very common gene fusion partner in TFE3 rearrangement-associated perivascular epithelioid cell tumors (PEComas) and melanotic Xp11 translocation renal cancers: Clinicopathologic, immunohistochemical, and molecular characteristics suggesting classification as a distinct entity. Am. J. Surg. Pathol. 2015, 39, 1181–1196. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Zhong, M.; Reuter, V.E.; Fallon, J.T.; Epstein, J.I.; Netto, G.J.; Antonescu, C.R. TFE3-fusion variant analysis defines specific clinicopathologic associations among Xp11 translocation cancers. Am. J. Surg. Pathol. 2016, 40, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.L.; Lazar, A.J. Applications of molecular testing to differential diagnosis. In Practical Soft Tissue Pathology: A Diagnostic Approach, 2nd ed.; Hornick, J.L., Ed.; Elsevier: Philadelphia, PA, USA, 2019; pp. 513–551. [Google Scholar]
- Alaggio, R.; Creytens, D. Myxoid pleomorphic liposarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 34–35. [Google Scholar]
- Sbaraglia, M.; Dei Tos, A.P.; Pedeutour, F. Atypical lipomatous tumour/well-differentiated liposarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 36–38. [Google Scholar]
- Evans, H.L. Atypical lipomatous tumor, its variants, and its combined forms: A study of 61 cases, with a minimum follow-up of 10 years. Am. J. Surg. Pathol. 2007, 31, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kraus, M.D.; Guillou, L.; Fletcher, C.D. Well-differentiated inflammatory liposarcoma: An uncommon and easily overlooked variant of a common sarcoma. Am. J. Surg. Pathol. 1997, 21, 518–527. [Google Scholar] [CrossRef]
- Clay, M.R.; Martinez, A.P.; Weiss, S.W.; Edgar, M.A. MDM2 and CDK4 immunohistochemistry: Should it be used in problematic differentiated lipomatous tumors? A new perspective. Am. J. Surg. Pathol. 2016, 40, 1647–1652. [Google Scholar] [CrossRef]
- Macarenco, R.S.; Erickson-Johnson, M.; Wang, X.; Folpe, A.A.; Rubin, B.P.; Nascimento, A.G.; Oliveira, A.M. Retroperitoneal lipomatous tumors without cytologic atypia: Are they lipomas? A clinicopathologic and molecular study of 19 cases. Am. J. Surg. Pathol. 2009, 33, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Creytens, D.; Marino-Enriquez, A. Atypical spindle cell/pleomorphic lipomatous tumour. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 34–35. [Google Scholar]
- Creytens, D.; Ferdinande, L. Diagnostic utility of STAT6 Immunohistochemistry in the diagnosis of fat-forming solitary fibrous tumors. Appl. Immunohistochem. Mol. Morphol. 2016, 24, e12–e13. [Google Scholar] [CrossRef] [PubMed]
- Dei Tos, A.P.; Marino-Enriquez, A.; Pedeutour, F. Dedifferentiated liposarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification Of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 39–41. [Google Scholar]
- McCormick, D.; Mentzel, T.; Beham, A.; Fletcher, C.D. Dedifferentiated liposarcoma. Clinicopathologic analysis of 32 cases suggesting a better prognostic subgroup among pleomorphic sarcomas. Am. J. Surg. Pathol. 1994, 18, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Henricks, W.H.; Chu, Y.C.; Goldblum, J.R.; Weiss, S.W. Dedifferentiated liposarcoma: A clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am. J. Surg. Pathol. 1997, 21, 271–281. [Google Scholar] [CrossRef]
- Italiano, A.; Bianchini, L.; Gjernes, E.; Keslair, F.; Ranchere-Vince, D.; Dumollard, J.M.; Haudebourg, J.; Leroux, A.; Mainguené, C.; Mainguené, C.; et al. Clinical and biological significance of CDK4 amplification in well-differentiated and dedifferentiated liposarcomas. Clin. Cancer Res. 2009, 15, 5696–5703. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.W.; Rao, V.K. Well-differentiated liposarcoma (atypical lipoma) of deep soft tissue of the extremities, retroperitoneum, and miscellaneous sites. A follow-up study of 92 cases with analysis of the incidence of “dedifferentiation”. Am. J. Surg. Pathol. 1992, 16, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.L.; Khurana, K.K.; Kemp, B.L.; Ayala, A.G. Heterologous elements in the dedifferentiated component of dedifferentiated liposarcoma. Am. J. Surg. Pathol. 1994, 18, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.G.; Kurtin, P.J.; Guillou, L.; Fletcher, C.D. Dedifferentiated liposarcoma: A report of nine cases with a peculiar neurallike whorling pattern associated with metaplastic bone formation. Am. J. Surg. Pathol. 1998, 22, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Mariño-Enríquez, A.; Fletcher, C.D.; Dal Cin, P.; Hornick, J.L. Dedifferentiated liposarcoma with “homologous” lipoblastic (pleomorphic liposarcoma-like) differentiation: Clinicopathologic and molecular analysis of a series suggesting revised diagnostic criteria. Am. J. Surg. Pathol. 2010, 34, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Gronchi, A.; Collini, P.; Miceli, R.; Valeri, B.; Renne, S.L.; Dagrada, G.; Fiore, M.; Sanfilippo, R.; Barisella, M.; Colombo, C.; et al. Myogenic differentiation and histologic grading are major prognostic determinants in retroperitoneal liposarcoma. Am. J. Surg. Pathol. 2015, 39, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Makise, N.; Sekimizu, M.; Kubo, T.; Wakai, S.; Hiraoka, N.; Komiyama, M.; Fukayama, M.; Kawai, A.; Ichikawa, H.; Yoshida, A. Clarifying the distinction between malignant peripheral nerve sheath tumor and dedifferentiated liposarcoma: A critical appraisal of the diagnostic utility of MDM and H3K27me3 status. Am. J. Surg. Pathol. 2018, 42, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Coindre, J.M.; Mariani, O.; Chibon, F.; Mairal, A.; De Saint Aubain Somerhausen, N.; Favre-Guillevin, E.; Bui, N.B.; Stoeckle, E.; Hostein, I.; Aurias, A. Most malignant fibrous histiocytomas developed in the retroperitoneum are dedifferentiated liposarcomas: A review of 25 cases initially diagnosed as malignant fibrous histiocytoma. Mod. Pathol. 2003, 16, 256–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedeutour, F.; Montgomery, E.A. Pleomorphic liposarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 45–46. [Google Scholar]
- Downes, K.A.; Goldblum, J.R.; Montgomery, E.A.; Fisher, C. Pleomorphic liposarcoma: A clinicopathologic analysis of 19 cases. Mod. Pathol. 2001, 14, 179–184. [Google Scholar] [CrossRef]
- Hornick, J.L.; Bosenberg, M.W.; Mentzel, T.; McMenamin, M.E.; Oliveira, A.M.; Fletcher, C.D. Pleomorphic liposarcoma: Clinicopathologic analysis of 57 cases. Am. J. Surg. Pathol. 2004, 28, 1257–1267. [Google Scholar] [CrossRef]
- Gebhard, S.; Coindre, J.M.; Michels, J.J.; Terrier, P.; Bertrand, G.; Trassard, M.; Taylor, S.; Château, M.C.; Marquès, B.; Picot, V.; et al. Pleomorphic liposarcoma: Clinicopathologic, immunohistochemical, and follow-up analysis of 63 cases: A study from the French Federation of Cancer Centers Sarcoma Group. Am. J. Surg. Pathol. 2002, 26, 601–616. [Google Scholar] [CrossRef]
- Fritz, B.; Schubert, F.; Wrobel, G.; Schwaenen, C.; Wessendorf, S.; Nessling, M.; Korz, C.; Rieker, R.J.; Montgomery, K.; Kucherlapati, R. Microarray-based copy number and expression profiling in dedifferentiated and pleomorphic liposarcoma. Cancer Res. 2002, 62, 2993–2998. [Google Scholar] [PubMed]
- Idbaih, A.; Coindre, J.M.; Derré, J.; Mariani, O.; Terrier, P.; Ranchère, D.; Mairal, A.; Aurias, A. Myxoid malignant fibrous histiocytoma and pleomorphic liposarcoma share very similar genomic imbalances. Lab. Investig. 2005, 85, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Taylor, B.S.; Banerji, S.; Ramos, A.H.; Lagos-Quintana, M.; Decarolis, P.L.; Shah, K.; Socci, N.D.; Weir, B.A.; Ho, A.; et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 2010, 42, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, W.J.; Jo, V.Y. Pleomorphic liposarcoma: Updates and current differential diagnosis. Semin. Diagn. Pathol. 2019, 36, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Antonescu, C.R. Epithelioid variant of pleomorphic liposarcoma: A comparative immunohistochemical and ultrastructural analysis of six cases with emphasis on overlapping features with epithelial malignancies. Ultrastruct. Pathol. 2002, 26, 299–308. [Google Scholar] [CrossRef]
- Goldblum, J.R.; Folpe, A.L.; Weiss, S.W. Liposarcoma. In Enzinger and Weiss’s Soft Tissue Tumors, 7th ed.; Elsevier: Philadelphia, PA, USA, 2020; pp. 520–522. [Google Scholar]
- Thway, K.; Nielsen, T.O. Myxoid liposarcoma. In WHO Classification of Tumours. Soft TISSUE and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 42–44. [Google Scholar]
- De Vreeze, R.S.; de Jong, D.; Tielen, I.H.; Ruijter, H.J.; Nederlof, P.M.; Haas, R.L.; van Coevorden, F. Primary retroperitoneal myxoid/round cell liposarcoma is a nonexisting disease: An immunohistochemical and molecular biological analysis. Mod. Pathol. 2009, 22, 223–231. [Google Scholar] [CrossRef]
- Setsu, N.; Miyake, M.; Wakai, S.; Nakatani, F.; Kobayashi, E.; Chuman, H.; Hiraoka, N.; Kawai, A.; Yoshida, A. Primary retroperitoneal myxoid liposarcomas. Am. J. Surg. Pathol. 2016, 40, 1286–1290. [Google Scholar] [CrossRef] [Green Version]
- Powers, M.P.; Wang, W.L.; Hernandez, V.S.; Patel, K.S.; Lev, D.C.; Lazar, A.J.; López-Terrada, D.H. Detection of myxoid liposarcoma-associated rearrangement variants including a newly identified breakpoint using an optimized RT-PCR assay. Mod. Pathol. 2010, 23, 1307–1315. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.A.; Easley, K.A.; Goldblum, J.R. Myxoid/round cell liposarcoma of the extremities. A clinicopathologic study of 29 cases with particular attention to extent of round cell liposarcoma. Am. J. Surg. Pathol. 1996, 20, 171–180. [Google Scholar] [CrossRef]
- Moreau, L.C.; Turcotte, R.; Ferguson, P.; Wunder, J.; Clarkson, P.; Masri, B.; Isler, M.; Dion, N.; Werier, J.; Ghert, M.; et al. Canadian Orthopaedic Oncology Society(CANOOS). Myxoid\round cell liposarcoma (MRCLS) revisited: An analysis of 418 primarily managed cases. Ann. Surg. Oncol. 2012, 19, 1081–1088. [Google Scholar] [CrossRef]
- Baranov, E.; McBride, M.J.; Bellizzi, A.M.; Ligon, A.H.; Fletcher, C.D.M.; Kadoch, C.; Hornick, J.L. A novel SS18-SSX fusion-specific antibody for the diagnosis of synovial sarcoma. Am. J. Surg. Pathol. 2020, 44, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Dry, S.M.; Fröhling, S. Leiomyosacroma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 195–197. [Google Scholar]
- Wile, A.G.; Evans, H.L.; Romsdahl, M.M. Leiomyosarcoma of soft tissue: A clinicopathologic study. Cancer 1981, 48, 1022–1032. [Google Scholar] [CrossRef]
- Shmookler, B.M.; Lauer, D.H. Retroperitoneal leiomyosarcoma. A clinicopathologic analysis of 36 cases. Am. J. Surg. Pathol. 1983, 7, 269–280. [Google Scholar] [PubMed]
- Farshid, G.; Pradhan, M.; Goldblum, J.; Weiss, S.W. Leiomyosarcoma of somatic soft tissues: A tumor of vascular origin with multivariate analysis of outcome in 42 cases. Am. J. Surg. Pathol. 2002, 26, 14–24. [Google Scholar] [CrossRef]
- Fletcher, C.D.M.; Mertens, F. Inflammatory leiomyosarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 193–194. [Google Scholar]
- Watanabe, R.; Schafernak, K.T.; Soares, F.A. EBV-associated smooth muscle tumour. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 190–192. [Google Scholar]
- Rubin, B.P.; Fletcher, C.D. Myxoid leiomyosarcoma of soft tissue, an underrecognized variant. Am. J. Surg. Pathol. 2000, 24, 927–936. [Google Scholar] [CrossRef]
- Yamamoto, T.; Minami, R.; Ohbayashi, C.; Inaba, M. Epithelioid leiomyosarcoma of the external deep soft tissue. Arch. Pathol. Lab. Med. 2002, 126, 468–470. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Miyajima, K.; Kawaguchi, K.; Tamiya, S.; Oshiro, Y.; Hachitanda, Y.; Oya, M.; Iwamoto, Y.; Tsuneyoshi, M. Pleomorphic leiomyosarcoma: Clinicopathologic and immunohistochemical study with special emphasis on its distinction from ordinary leiomyosarcoma and malignant fibrous histiocytoma. Am. J. Surg. Pathol. 2001, 25, 1030–1038. [Google Scholar] [CrossRef]
- Chen, E.; O’Connell, F.; Fletcher, C.D. Dedifferentiated leiomyosarcoma: Clinicopathological analysis of 18 cases. Histopathology 2011, 59, 1135–1143. [Google Scholar] [CrossRef]
- Hornick, J.L.; Fletcher, C.D. Criteria for malignancy in nonvisceral smooth muscle tumors. Ann. Diagn. Pathol. 2003, 7, 60–66. [Google Scholar] [CrossRef]
- Weiss, S.W. Smooth muscle tumors of soft tissue. Adv. Anat. Pathol. 2002, 9, 351–359. [Google Scholar] [CrossRef]
- Cyril, F.; Montgomery, E.A.; Thway, K. Biopsy Interpretation of Soft Tissue Tumors, 2nd ed.; Wolters Kluwer: Philadelphia, PA, USA, 2016; pp. 134–135. [Google Scholar]
- Iwata, J.; Fletcher, C.D. Immunohistochemical detection of cytokeratin and epithelial membrane antigen in leiomyosarcoma: A systematic study of 100 cases. Pathol. Int. 2000, 50, 7–14. [Google Scholar] [CrossRef]
- Demicco, E.G.; Fritchie, K.J.; Han, A. Solitary fibrous tumour. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 104–108. [Google Scholar]
- Yamada, K.; Abiko, K.; Kido, A.; Minamiguchi, S.; Horie, A.; Mandai, M. Solitary fibrous tumor arising from pelvic retroperitoneum: A report of two cases and a review of the literature. J. Obstet. Gynaecol. Res. 2019, 45, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chu, X.; Yi, Y.; Tong, L.; Dai, Y. Malignant solitary fibrous tumor in retroperitoneum: A case report and literature review. Medicine 2017, 96, e6373. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Crago, A.M.; Rosenberg, M.; O’Connor, R.; Walker, S.R.; Ambrogio, L.; Auclair, D.; McKenna, A.; Heinrich, M.C.; Frank, D.A.; et al. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat. Genet. 2013, 45, 131–132. [Google Scholar] [CrossRef] [Green Version]
- De Saint Aubain Somerhausen, N.; Rubin, B.P.; Fletcher, C.D. Myxoid solitary fibrous tumor: A study of seven cases with emphasis on differential diagnosis. Mod. Pathol. 1999, 12, 463–471. [Google Scholar] [PubMed]
- Guillou, L.; Gebhard, S.; Coindre, J.M. Lipomatous hemangiopericytoma: A fat-containing variant of solitary fibrous tumor? Clinicopathologic, immunohistochemical, and ultrastructural analysis of a series in favor of a unifying concept. Hum. Pathol. 2000, 31, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Guillou, L.; Gebhard, S.; Coindre, J.M. Orbital and extraorbital giant cell angiofibroma: A giant cell-rich variant of solitary fibrous tumor? Clinicopathologic and immunohistochemical analysis of a series in favor of a unifying concept. Am. J. Surg. Pathol. 2000, 24, 971–979. [Google Scholar] [CrossRef]
- Olson, N.J.; Linos, K. Dedifferentiated solitary fibrous tumor: A concise review. Arch. Pathol. Lab. Med. 2018, 142, 761–766. [Google Scholar] [CrossRef] [Green Version]
- Demicco, E.G.; Wagner, M.J.; Maki, R.G.; Gupta, V.; Iofin, I.; Lazar, A.J.; Wang, W.L. Risk assessment in solitary fibrous tumors: Validation and refinement of a risk stratification model. Mod. Pathol. 2017, 30, 1433–1442. [Google Scholar] [CrossRef]
- Creytens, D.; Ferdinande, L.; Van Dorpe, J. multifocal cytokeratin expression in a dedifferentiated solitary fibrous tumor with heterologous rhabdomyosarcomatous differentiation: A challenging diagnosis! Int. J. Surg. Pathol. 2018, 26, 423–427. [Google Scholar] [CrossRef]
- Doyle, L.A.; Vivero, M.; Fletcher, C.D.; Mertens, F.; Hornick, J.L. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod. Pathol. 2014, 27, 390–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, L.A.; Tao, D.; Mariño-Enríquez, A. STAT6 is amplified in a subset of dedifferentiated liposarcoma. Mod. Pathol. 2014, 27, 1231–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creytens, D.; Libbrecht, L.; Ferdinande, L. Nuclear expression of STAT6 in dedifferentiated liposarcomas with a solitary fibrous tumor-like morphology: A diagnostic pitfall. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Creytens, D. Malignant solitary fibrous tumour of the kidney with lymph node and liver metastases: Beware of STAT6 expression in dedifferentiated liposarcoma with a solitary fibrous tumour-like morphology. Pathology 2017, 49, 671. [Google Scholar] [CrossRef]
- Yamamoto, H. Inflammatory myofibroblastic tumour. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 109–111. [Google Scholar]
- Coffin, C.M.; Watterson, J.; Priest, J.R.; Dehner, L.P. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am. J. Surg. Pathol. 1995, 19, 859–872. [Google Scholar] [CrossRef]
- Koirala, R.; Shakya, V.C.; Agrawal, C.S.; Khaniya, S.; Pandey, S.R.; Adhikary, S.; Pathania, O.P. Retroperitoneal inflammatory myofibroblastic tumor. Am. J. Surg. 2010, 199, e17–e19. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Suurmeijer, A.J.; Zhang, L.; Sung, Y.S.; Jungbluth, A.A.; Travis, W.D.; Al-Ahmadie, H.; Fletcher, C.D.; Alaggio, R. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am. J. Surg. Pathol. 2015, 39, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Alassiri, A.H.; Ali, R.H.; Shen, Y.; Lum, A.; Strahlendorf, C.; Deyell, R.; Rassekh, R.; Sorensen, P.H.; Laskin, J.; Marra, M.; et al. ETV6-NTRK3 is expressed in a subset of ALK-negative inflammatory myofibroblastic tumors. Am. J. Surg. Pathol. 2016, 40, 1051–1061. [Google Scholar] [CrossRef]
- Coffin, C.M.; Hornick, J.L.; Fletcher, C.D. Inflammatory myofibroblastic tumor: Comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am. J. Surg. Pathol. 2007, 31, 509–520. [Google Scholar] [CrossRef]
- Mariño-Enríquez, A.; Wang, W.L.; Roy, A.; Lopez-Terrada, D.; Lazar, A.J.; Fletcher, C.D.; Coffin, C.M.; Hornick, J.L. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am. J. Surg. Pathol. 2011, 35, 135–144. [Google Scholar] [CrossRef]
- Zen, Y.; Onodera, M.; Inoue, D.; Kitao, A.; Matsui, O.; Nohara, T.; Namiki, M.; Kasashima, S.; Kawashima, A.; Matsumoto, Y.; et al. Retroperitoneal fibrosis: A clinicopathologic study with respect to immunoglobulin G4. Am. J. Surg. Pathol. 2009, 33, 1833–1839. [Google Scholar] [CrossRef] [PubMed]
- Rudzinski, E.R. Embryonal rhabdomyosarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 201–204. [Google Scholar]
- Crist, W.M.; Raney, R.B.; Tefft, M.; Heyn, R.; Hays, D.M.; Newton, W.; Beltangady, M.; Maurer, H.M. Soft tissue sarcomas arising in the retroperitoneal space in children. A report from the Intergroup Rhabdomyosarcoma Study (IRS) Committee. Cancer 1985, 56, 2125–2132. [Google Scholar] [CrossRef]
- Furlong, M.A.; Mentzel, T.; Fanburg-Smith, J.C. Pleomorphic rhabdomyosarcoma in adults: A clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod. Pathol. 2001, 14, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.F.; Fletcher, C.D. Spindle cell rhabdomyosarcoma in adults. Am. J. Surg. Pathol. 2005, 29, 1106–1113. [Google Scholar]
- Qualman, S.; Lynch, J.; Bridge, J.; Parham, D.; Teot, L.; Meyer, W.; Pappo, A. Prevalence and clinical impact of anaplasia in childhood rhabdomyosarcoma: A report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Cancer 2008, 113, 3242–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudzinski, E.R.; Teot, L.A.; Anderson, J.R.; Moore, J.; Bridge, J.A.; Barr, F.G.; Gastier-Foster, J.M.; Skapek, S.X.; Hawkins, D.S.; Parham, D.M. Dense pattern of embryonal rhabdomyosarcoma, a lesion easily confused with alveolar rhabdomyosarcoma: A report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Am. J. Clin. Pathol. 2013, 140, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Kohashi, K.; Bode-Lesniewska, B. Alveolar rhabdomyosarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 205–208. [Google Scholar]
- Montgomery, E.A.; Dry, S.M. Pleomorphic rhabdomyosarcoma. In WHO Classification of Tumours. Soft Tissue and Bone tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 209–210. [Google Scholar]
- Agaram, N.P.; Szuhai, K. Spindle cell/sclerosing rhabdomyosarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 211–213. [Google Scholar]
- Trombatore, C.; Rosario, C.; Giovanni, L.D.; Gaetano, M.; Giuseppe, P.; Antonio, D.C. Dedifferentiated liposarcoma of retroperitoneum with extensive osteosarcomatous component. Int. Surg. 2016, 101, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.L.; Antonescu, C.R.; Bahrami, A. Infantile fibrosarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 119–121. [Google Scholar]
- Nielsen, G.P.; Chi, P. Malignant peripheral nerve sheath tumour. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 254–257. [Google Scholar]
- Ducatman, B.S.; Scheithauer, B.W.; Piepgras, D.G.; Reiman, H.M.; Ilstrup, D.M. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986, 57, 2006–2021. [Google Scholar] [CrossRef]
- Kourea, H.P.; Bilsky, M.H.; Leung, D.H.; Lewis, J.J.; Woodruff, J.M. Subdiaphragmatic and intrathoracic paraspinal malignant peripheral nerve sheath tumors: A clinicopathologic study of 25 patients and 26 tumors. Cancer 1998, 82, 2191–2203. [Google Scholar] [CrossRef]
- McMenamin, M.E.; Fletcher, C.D. Expanding the spectrum of malignant change in schwannomas: Epithelioid malignant change, epithelioid malignant peripheral nerve sheath tumor, and epithelioid angiosarcoma: A study of 17 cases. Am. J. Surg. Pathol. 2001, 25, 13–25. [Google Scholar] [CrossRef]
- Ghali, V.S.; Gold, J.E.; Vincent, R.A.; Cosgrove, J.M. Malignant peripheral nerve sheath tumor arising spontaneously from retroperitoneal ganglioneuroma: A case report, review of the literature, and immunohistochemical study. Hum. Pathol. 1992, 23, 72–75. [Google Scholar] [CrossRef]
- Van Haverbeke, C.; Ferdinande, L.; Verbeke, S.; Van Dorpe, J.; Creytens, D. Malignant peripheral nerve sheath tumour with heterologous liposarcomatous differentiation: Case report and review of the literature. Pathology 2018, 50, 475–478. [Google Scholar] [CrossRef]
- Laskin, W.B.; Weiss, S.W.; Bratthauer, G.L. Epithelioid variant of malignant peripheral nerve sheath tumor (malignant epithelioid schwannoma). Am. J. Surg. Pathol. 1991, 15, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.M.; Kim, A.; Lazar, A.J.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1—A consensus overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Fletcher, C.D.; Hornick, J.L. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod. Pathol. 2016, 29, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Hornick, J.L.; Nielsen, G.P. Beyond “Triton”: Malignant peripheral nerve sheath tumors with complete heterologous rhabdomyoblastic differentiation mimicking spindle cell rhabdomyosarcoma. Am. J. Surg. Pathol. 2019, 43, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Hameed, M. Extraskeletal osteosarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 224–225. [Google Scholar]
- Orta, L.; Suprun, U.; Goldfarb, A.; Bleiweiss, I.; Jaffer, S. Radiation-associated extraskeletal osteosarcoma of the chest wall. Arch. Pathol. Lab. Med. 2006, 130, 198–200. [Google Scholar] [CrossRef]
- Lidang Jensen, M.; Schumacher, B.; Myhre Jensen, O.; Steen Nielsen, O.; Keller, J. Extraskeletal osteosarcomas: A clinicopathologic study of 25 cases. Am. J. Surg. Pathol. 1998, 22, 588–594. [Google Scholar] [CrossRef]
- Bane, B.L.; Evans, H.L.; Ro, J.Y.; Carrasco, C.H.; Grignon, D.J.; Benjamin, R.S.; Ayala, A.G. Extraskeletal osteosarcoma. A clinicopathologic review of 26 cases. Cancer 1990, 65, 2762–2770. [Google Scholar] [CrossRef]
- Yamashita, K.; Kohashi, K.; Yamada, Y.; Nishida, Y.; Urakawa, H.; Oda, Y.; Toyokuni, S. Primary extraskeletal osteosarcoma: A clinicopathological study of 18 cases focusing on MDM2 amplification status. Hum. Pathol. 2017, 63, 63–69. [Google Scholar] [CrossRef]
- Conner, J.R.; Hornick, J.L. SATB2 is a novel marker of osteoblastic differentiation in bone and soft tissue tumours. Histopathology 2013, 63, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Machado, I.; Navarro, S.; Picci, P.; Llombart-Bosch, A. The utility of SATB2 immunohistochemical expression in distinguishing between osteosarcomas and their malignant bone tumor mimickers, such as Ewing sarcomas and chondrosarcomas. Pathol. Res. Pract. 2016, 212, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.M.; Weiss, S.W.; Folpe, A.L. Heterotopic mesenteric ossification: A distinctive pseudosarcoma commonly associated with intestinal obstruction. Am. J. Surg. Pathol. 2006, 30, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Magro, G.; Salvatorelli, L.; Puzzo, L.; Musumeci, G.; Bisceglia, M.; Parenti, R. Oncofetal expression of Wilms’ tumor 1 (WT1) protein in human fetal, adult and neoplastic skeletal muscle tissues. Acta Histochem. 2015, 117, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Winnepenninckx, V.; De Vos, R.; Debiec-Rychter, M.; Samson, I.; Brys, P.; Hagemeijer, A.; Sciot, R. Calcifying/ossifying synovial sarcoma shows t(X;18) with SSX2 involvement and mitochondrial calcifications. Histopathology 2001, 38, 141–145. [Google Scholar] [CrossRef]
- Suurmeijer, A.J.H.; Ladanyi, M.; Nielsen, T.O. Synovial sarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 290–293. [Google Scholar]
- Sultan, I.; Rodriguez-Galindo, C.; Saab, R.; Yasir, S.; Casanova, M.; Ferrari, A. Comparing children and adults with synovial sarcoma in the surveillance, epidemiology, and end results program, 1983 to 2005: An analysis of 1268 patients. Cancer 2009, 115, 3537–3547. [Google Scholar] [CrossRef]
- Makhlouf, H.R.; Ahrens, W.; Agarwal, B.; Dow, N.; Marshalleck, J.J.; Lee, E.L.; Dotto, J.E.; Hui, P.; Sobin, L.H.; Oliveira, A.; et al. Synovial sarcoma of the stomach: A clinicopathologic, immunohistochemical, and molecular genetic study of 10 cases. Am. J. Surg. Pathol. 2008, 32, 275–281. [Google Scholar] [CrossRef]
- Schoolmeester, J.K.; Cheville, J.C.; Folpe, A.L. Synovial sarcoma of the kidney: A clinicopathologic, immunohistochemical, and molecular genetic study of 16 cases. Am. J. Surg. Pathol. 2014, 38, 60–65. [Google Scholar] [CrossRef]
- Fisher, C.; Folpe, A.L.; Hashimoto, H.; Weiss, S.W. Intra-abdominal synovial sarcoma: A clinicopathological study. Histopathology 2004, 45, 245–253. [Google Scholar] [CrossRef]
- Van de Rijn, M.; Barr, F.G.; Xiong, Q.B.; Hedges, M.; Shipley, J.; Fisher, C. Poorly differentiated synovial sarcoma: An analysis of clinical, pathologic, and molecular genetic features. Am. J. Surg. Pathol. 1999, 23, 106–112. [Google Scholar] [CrossRef]
- Foo, W.C.; Cruise, M.W.; Wick, M.R.; Hornick, J.L. Immunohistochemical staining for TLE1 distinguishes synovial sarcoma from histologic mimics. Am. J. Clin. Pathol. 2011, 135, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Amary, M.F.; Berisha, F.; Bernardi Fdel, C.; Herbert, A.; James, M.; Reis-Filho, J.S.; Fisher, C.; Nicholson, A.G.; Tirabosco, R.; Diss, T.C.; et al. Detection of SS18-SSX fusion transcripts in formalin-fixed paraffin-embedded neoplasms: Analysis of conventional RT-PCR, qRT-PCR and dual color FISH as diagnostic tools for synovial sarcoma. Mod. Pathol. 2007, 20, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.P.; Antonescu, C.R.; Ladanyi, M. Desmoplasic small round cell tumours. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 306–308. [Google Scholar]
- Gerald, W.; Rosai, J. Case 2. Desmoplastic small cell tumor with divergent differentiation. Pediatr. Pathol. 1989, 9, 177–183. [Google Scholar] [CrossRef]
- Ordóñez, N.G. Desmoplastic small round cell tumor: I: A histopathologic study of 39 cases with emphasis on unusual histological patterns. Am. J. Surg. Pathol. 1998, 22, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Lae, M.E.; Roche, P.C.; Jin, L.; Lloyd, R.V.; Nascimento, A.G. Desmoplastic small round cell tumor: A clinicopathologic, immunohistochemical, and molecular study of 32 tumors. Am. J. Surg. Pathol. 2002, 26, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Lamhamedi-Cherradi, S.E.; Cuglievan, B.; Menegaz, B.A.; Camacho, P.; Huh, W.; Ramamoorthy, V.; Anderson, P.M.; Pollock, R.E.; Lev, D.C.; et al. Multimodality treatment of desmoplastic small round cell tumor: Chemotherapy and complete cytoreductive surgery improve patient survival. Clin. Cancer Res. 2018, 24, 4865–4873. [Google Scholar] [CrossRef] [Green Version]
- Pasquinelli, G.; Montanaro, L.; Martinelli, G.N. Desmoplastic small round-cell tumor: A case report on the large cell variant with immunohistochemical, ultrastructural, and molecular genetic analysis. Ultrastruct. Pathol. 2000, 24, 333–337. [Google Scholar] [CrossRef]
- Thway, K.; Noujaim, J.; Zaidi, S.; Miah, A.B.; Benson, C.; Messiou, C.; Jones, R.L.; Fisher, C. Desmoplastic small round cell tumor: Pathology, genetics, and potential therapeutic strategies. Int. J. Surg. Pathol. 2016, 24, 672–684. [Google Scholar] [CrossRef]
- Barnoud, R.; Sabourin, J.C.; Pasquier, D.; Ranchère, D.; Bailly, C.; Terrier-Lacombe, M.J.; Pasquier, B. Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: A comparative study with other small round cell tumors. Am. J. Surg. Pathol. 2000, 24, 830–836. [Google Scholar] [CrossRef]
- Hung, Y.P.; Lee, J.P.; Bellizzi, A.M.; Hornick, J.L. PHOX2B reliably distinguishes neuroblastoma among small round blue cell tumours. Histopathology 2017, 71, 786–794. [Google Scholar] [CrossRef]
- Doyle, L.A.; Argani, P.; Hornick, J.L. PEComa. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 312–314. [Google Scholar]
- Folpe, A.L.; Mentzel, T.; Lehr, H.A.; Fisher, C.; Balzer, B.L.; Weiss, S.W. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: A clinicopathologic study of 26 cases and review of the literature. Am. J. Surg. Pathol. 2005, 29, 1558–1575. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Hornick, J.L.; Fletcher, C.D. PEComa of the gastrointestinal tract: Clinicopathologic study of 35 cases with evaluation of prognostic parameters. Am. J. Surg. Pathol. 2013, 37, 1769–1782. [Google Scholar] [CrossRef] [PubMed]
- Hornick, J.L.; Fletcher, C.D. Sclerosing PEComa: Clinicopathologic analysis of a distinctive variant with a predilection for the retroperitoneum. Am. J. Surg. Pathol. 2008, 32, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Larque, A.B.; Kradin, R.L.; Chebib, I.; Nielsen, G.P.; Selig, M.K.; Thiele, E.A.; Stemmer-Rachamimov, A.; Bredella, M.A.; Kurzawa, P.; Deshpande, V. Fibroma-like PEComa: A tuberous sclerosis complex-related lesion. Am. J. Surg. Pathol. 2018, 42, 500–505. [Google Scholar] [CrossRef]
- Folpe, A.L.; Kwiatkowski, D.J. Perivascular epithelioid cell neoplasms: Pathology and pathogenesis. Hum. Pathol. 2010, 41, 1–15. [Google Scholar] [CrossRef]
- Argani, P.; Aulmann, S.; Illei, P.B.; Netto, G.J.; Ro, J.; Cho, H.Y.; Dogan, S.; Ladanyi, M.; Martignoni, G.; Goldblum, J.R.; et al. A distinctive subset of PEComas harbors TFE3 gene fusions. Am. J. Surg. Pathol. 2010, 34, 1395–1406. [Google Scholar] [CrossRef]
- Dei Tos, A.P.; Mertens, F.; Pillay, N. Undifferentiated sarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 318–320. [Google Scholar]
- Fletcher, C.D. Undifferentiated sarcomas: What to do? And does it matter? A surgical pathology perspective. Ultrastruct. Pathol. 2008, 32, 31–36. [Google Scholar] [CrossRef]
- Laskin, W.B.; Silverman, T.A.; Enzinger, F.M. Postradiation soft tissue sarcomas. An analysis of 53 cases. Cancer 1988, 62, 2330–2340. [Google Scholar] [CrossRef]
- Fletcher, C.D.; Gustafson, P.; Rydholm, A.; Willén, H.; Akerman, M. Clinicopathologic re-evaluation of 100 malignant fibrous histiocytomas: Prognostic relevance of subclassification. J. Clin. Oncol. 2001, 19, 3045–3050. [Google Scholar] [CrossRef]
- Deyrup, A.T.; Haydon, R.C.; Huo, D.; Ishikawa, A.; Peabody, T.D.; He, T.C.; Montag, A.G. Myoid differentiation and prognosis in adult pleomorphic sarcomas of the extremity: An analysis of 92 cases. Cancer 2003, 98, 805–813. [Google Scholar] [CrossRef]
- Chung, L.; Lau, S.K.; Jiang, Z.; Loera, S.; Bedel, V.; Ji, J.; Weiss, L.M.; Chu, P.G. Overlapping features between dedifferentiated liposarcoma and undifferentiated high-grade pleomorphic sarcoma. Am. J. Surg. Pathol. 2009, 33, 1594–1600. [Google Scholar] [CrossRef] [PubMed]
- De Álava, E.; Lessnick, S.L.; Stamenkovic, I. Ewing sarcoma. In WHO Classification of Tumours. Soft tissue and bone tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 323–325. [Google Scholar]
- Rud, N.P.; Reiman, H.M.; Pritchard, D.J.; Frassica, F.J.; Smithson, W.A. Extraosseous Ewing’s sarcoma. A study of 42 cases. Cancer 1989, 64, 1548–1553. [Google Scholar] [CrossRef]
- Cash, T.; McIlvaine, E.; Krailo, M.D.; Lessnick, S.L.; Lawlor, E.R.; Laack, N.; Sorger, J.; Marina, N.; Grier, H.E.; Granowetter, L.; et al. Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2016, 63, 1771–1779. [Google Scholar] [CrossRef]
- Jimenez, R.E.; Folpe, A.L.; Lapham, R.L.; Ro, J.Y.; O’Shea, P.A.; Weiss, S.W.; Amin, M.B. Primary Ewing’s sarcoma/primitive neuroectodermal tumor of the kidney: A clinicopathologic and immunohistochemical analysis of 11 cases. Am. J. Surg. Pathol. 2002, 26, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Movahedi-Lankarani, S.; Hruban, R.H.; Westra, W.H.; Klimstra, D.S. Primitive neuroectodermal tumors of the pancreas: A report of seven cases of a rare neoplasm. Am. J. Surg. Pathol. 2002, 26, 1040–1047. [Google Scholar] [CrossRef]
- Ulusan, S.; Koc, Z.; Canpolat, E.T.; Colakoglu, T. Radiological findings of primary retroperitoneal Ewing sarcoma. Acta Radiol. 2007, 48, 814–818. [Google Scholar] [CrossRef]
- Spacek, J.; Kopeckova, K.; Kosina, J.; Pacovsky, J.; Petera, J.; Krbal, L.; Mrhalová, M.; Dvorak, P.; Broďák, M. Primary retroperitoneal Ewing’s sarcoma. Rozhl. Chir. 2019, 98, 121–124. [Google Scholar]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [Green Version]
- Llombart-Bosch, A.; Machado, I.; Navarro, S.; Bertoni, F.; Bacchini, P.; Alberghini, M.; Karzeladze, A.; Savelov, N.; Petrov, S.; Alvarado-Cabrero, I.; et al. Histological heterogeneity of Ewing’s sarcoma/PNET: An immunohistochemical analysis of 415 genetically confirmed cases with clinical support. Virchows Arch. 2009, 455, 397–411. [Google Scholar] [CrossRef]
- Machado, I.; Noguera, R.; Mateos, E.A.; Calabuig-Fariñas, S.; López, F.I.; Martínez, A.; Navarro, S.; Llombart-Bosch, A. The many faces of atypical Ewing’s sarcoma. A true entity mimicking sarcomas, carcinomas and lymphomas. Virchows Arch. 2011, 458, 281–290. [Google Scholar] [CrossRef]
- Bishop, J.A.; Alaggio, R.; Zhang, L.; Seethala, R.R.; Antonescu, C.R. Adamantinoma-like Ewing family tumors of the head and neck: A pitfall in the differential diagnosis of basaloid and myoepithelial carcinomas. Am. J. Surg. Pathol. 2015, 39, 1267–1274. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, A.; Sekine, S.; Tsuta, K.; Fukayama, M.; Furuta, K.; Tsuda, H. NKX2.2 is a useful immunohistochemical marker for Ewing sarcoma. Am. J. Surg. Pathol. 2012, 36, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.L.; Patel, N.R.; Caragea, M.; Hogendoorn, P.C.; López-Terrada, D.; Hornick, J.L.; Lazar, A.J. Expression of ERG, an Ets family transcription factor, identifies ERG-rearranged Ewing sarcoma. Mod. Pathol. 2012, 25, 1378–1383. [Google Scholar] [CrossRef] [PubMed]
- Magro, G.; Salvatorelli, L.; Alaggio, R.; D’Agata, V.; Nicoletti, F.; Di Cataldo, A.; Parenti, R. Diagnostic utility of cyclin D1 in the diagnosis of small round blue cell tumors in children and adolescents. Hum. Pathol. 2017, 60, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Yoshida, A. CIC-rearranged sarcoma. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 330–332. [Google Scholar]
- Antonescu, C.R.; Puls, F.; Tirode, F. Sarcoma with BCOR genetic alterations. In WHO Classification of Tumours. Soft Tissue and Bone Tumours, 5th ed.; The WHO Classification of Tumours Editorial Board, Ed.; IARC Press: Lyon, France, 2020; pp. 333–335. [Google Scholar]
- Mentzel, T.; Calonje, E.; Wadden, C.; Camplejohn, R.S.; Beham, A.; Smith, M.A.; Fletcher, C.D. Myxofibrosarcoma. Clinicopathologic analysis of 75 cases with emphasis on the low-grade variant. Am. J. Surg. Pathol. 1996, 20, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Rekhi, B.; Ingle, A.; Agarwal, M.; Puri, A.; Laskar, S.; Jambhekar, N.A. Alveolar soft part sarcoma ‘revisited’: Clinicopathological review of 47 cases from a tertiary cancer referral centre, including immunohistochemical expression of TFE3 in 22 cases and 21 other tumours. Pathology 2012, 44, 11–17. [Google Scholar] [CrossRef]
- Meis-Kindblom, J.M.; Kindblom, L.G. Angiosarcoma of soft tissue: A study of 80 cases. Am. J. Surg. Pathol. 1998, 22, 683–697. [Google Scholar] [CrossRef]
- Katabuchi, H.; Honda, R.; Tajima, T.; Ohtake, H.; Kageshita, T.; Ono, T.; Okamura, H. Clear cell sarcoma arising in the retroperitoneum. Int. J. Gynecol. Cancer 2002, 12, 124–127. [Google Scholar] [CrossRef]
- Fukuda, T.; Ishikawa, H.; Ohnishi, Y.; Tachikawa, S.; Onizuka, S.; Sakashita, I. Extraskeletal myxoid chondrosarcoma arising from the retroperitoneum. Am. J. Clin. Pathol. 1986, 85, 514–519. [Google Scholar] [CrossRef]
- Paal, E.; Miettinen, M. Retroperitoneal leiomyomas: A clinicopathologic and immunohistochemical study of 56 cases with a comparison to retroperitoneal leiomyosarcomas. Am. J. Surg. Pathol. 2001, 25, 1355–1363. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relatively Common Sarcomas | Rare Sarcomas |
|---|---|
| Liposarcoma (well-differentiated and dedifferentiated subtypes) Leiomyosarcoma | Solitary fibrous tumor (malignant) Inflammatory myofibroblastic tumor Rhabdomyosarcoma MPNST Extraskeletal osteosarcoma Synovial sarcoma Desmoplastic small round cell tumor PEComa (malignant) Undifferentiated pleomorphic sarcoma Extraskeletal Ewing sarcoma |
| Pattern | Tumor Types |
|---|---|
| Spindle cell | Leiomyosarcoma Solitary fibrous tumor Inflammatory myofibroblastic tumor MPNST Monophasic synovial sarcoma |
| Round cell | Poorly differentiated synovial sarcoma Desmoplastic small round cell tumor Extraskeletal Ewing sarcoma |
| Epithelioid cell | Epithelioid inflammatory myofibroblastic sarcoma Epithelioid MPNST PEComa |
| Pleomorphic cell | Dedifferentiated liposarcoma Pleomorphic leiomyosarcoma Pleomorphic rhabdomyosarcoma Undifferentiated pleomorphic sarcoma |
| Adipocytic component | Liposarcoma (well differentiated, dedifferentiated, myxoid, and pleomorphic subtype) Lipomatous (fat-forming) solitary fibrous tumor |
| Prominent inflammatory cells | Inflammatory well-differentiated liposarcoma Inflammatory myofibroblastic tumor |
| Tumor osteoid and bone | Extraskeletal osteosarcoma |
| Liposarcoma (Well-Differentiated/Dedifferentiated) | Solitary Fibrous Tumor | Inflammatory Myofibroblastic Tumor | Leiomyosarcoma | MPNST | Synovial Sarcoma | DSRCT | PEComa | Extraskeletal Ewing Sarcoma | |
|---|---|---|---|---|---|---|---|---|---|
| SMA | − (a) | +/− | +/− | + | − | − | − | + | − |
| Desmin | − (a) | − | +/− | + | − | − | + | +/− | − |
| CD34 | − | + | +/− | − | + | − | − | − | − |
| S100 protein | +/− | − | − | − | + | +/− | − | +/− | +/− |
| MDM2 | + | − | − | − | − (e) | − | − | − | − |
| CDK4 | + | − | − | − | − | − | − | − | − |
| STAT6 | − (b) | + (d) | − | − | − | − | − | − | − |
| ALK | − | − | + | − | − (f) | − | − | − | − |
| CD99 | − | + | − | − | − | + | + | − | + |
| H3K27me3 | Retained (c) | Retained | NA | Retained | Loss (g) | Retained (h) | NA | NA | Retained |
| NKX2.2 | − | − | − | − | − | − (i) | − (j) | − | + |
| TLE1 | − | +/− | − | − | +/− | + | − | − | − |
| SOX10 | − | − | − | − | + | − | − | − | − |
| WT1 | − | − | − | − | − | − | + | − | − |
| HMB-45, melan-A | − | − | − | − | − | − | − | + | − |
| Cytokeratin, EMA | − | +/− | +/− | +/− | − | + | + | − | +/− |
| Tumor Types | Cytogenetic Alterations | Molecular Alterations |
|---|---|---|
| Well-differentiated liposarcoma | Supernumerary ring or giant marker chromosome(s) | MDM2 amplification, other co-amplified genes CDK4, HMGA2, TSPAN31, YEATSA4, CPM, FRS2 |
| Dedifferentiated liposarcoma | Supernumerary ring or giant marker chromosome(s) | MDM2 amplification, other co-amplified genes CDK4, JUN, TERT, CPM, MAP3K5 |
| Myxoid liposarcoma | t(12;16)(q13;p11) t(12;22)(q13;q12) | FUS-DDIT3 EWSR1-DDIT3 |
| Solitary fibrous tumor | Inv(12)(q13q13) | NAB2-STAT6 fusion |
| Inflammatory myofibroblastic tumor | t(1;2)(q22;p23) t(2;19)(p23;p13) t(2;17)(p23;q23) | TPM3-ALK fusion TPM4-ALK fusion CLTC-ALK fusion ROS1 and PDGFRB rearrangement |
| Epithelioid inflammatory myofibroblastic sarcoma | t(2;2)(p23;q13) | RANBP2-ALK fusion |
| Embryonal rhabdomyosarcoma | Complex karyotypes; loss of heterozygosity at 11p15.5 | |
| Alveolar rhabdomyosarcoma | t(2;13)(q35;q14) t(1;13)(p36;q14) | PAX3-FOXO1A fusion PAX7-FOXO1A fusion |
| MPNST | Complex karyotypes; inactivation mutations in NF1, CDKN2A/CDKN2B, EED, or SUZ12 | |
| Epithelioid MPNST | SMARCB1 gene inactivation | |
| Synovial sarcoma | t(X;18)(p11;q11) | SS18-SSX1 fusion SS18-SSX2 fusion |
| Desmoplastic small round cell tumor | t(11;22)(p13;q12) | EWSR1-WT1, EWSR1-ERG, EWDR1-FLI1 fusion |
| PEComa | Deletion of 16p, the location of TSC2 gene | SFPQ-TFE3, DVL2-TFE3, NONO-TFE3 fusion |
| Extraskeletal Ewing sarcoma | t(11;22)(q24;q12) t(21;22)(q12;q12) t(2;22)(q33;q12) t(7;22)(p22;q12) t(17;22)(q12;q12) | EWSR1-FLI1 fusion EWSR1-ERG fusion EWSR1-FEV fusion EWSR1-ETV1 fusion EWSR1-E1AF fusion |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.H.; Ro, J.Y. Retroperitoneal Sarcomas: An Update on the Diagnostic Pathology Approach. Diagnostics 2020, 10, 642. https://doi.org/10.3390/diagnostics10090642
Choi JH, Ro JY. Retroperitoneal Sarcomas: An Update on the Diagnostic Pathology Approach. Diagnostics. 2020; 10(9):642. https://doi.org/10.3390/diagnostics10090642
Chicago/Turabian StyleChoi, Joon Hyuk, and Jae Y. Ro. 2020. "Retroperitoneal Sarcomas: An Update on the Diagnostic Pathology Approach" Diagnostics 10, no. 9: 642. https://doi.org/10.3390/diagnostics10090642
APA StyleChoi, J. H., & Ro, J. Y. (2020). Retroperitoneal Sarcomas: An Update on the Diagnostic Pathology Approach. Diagnostics, 10(9), 642. https://doi.org/10.3390/diagnostics10090642