
Update on Molecular Genetics of Gastrointestinal Stromal Tumors


Abstract
1. Introduction
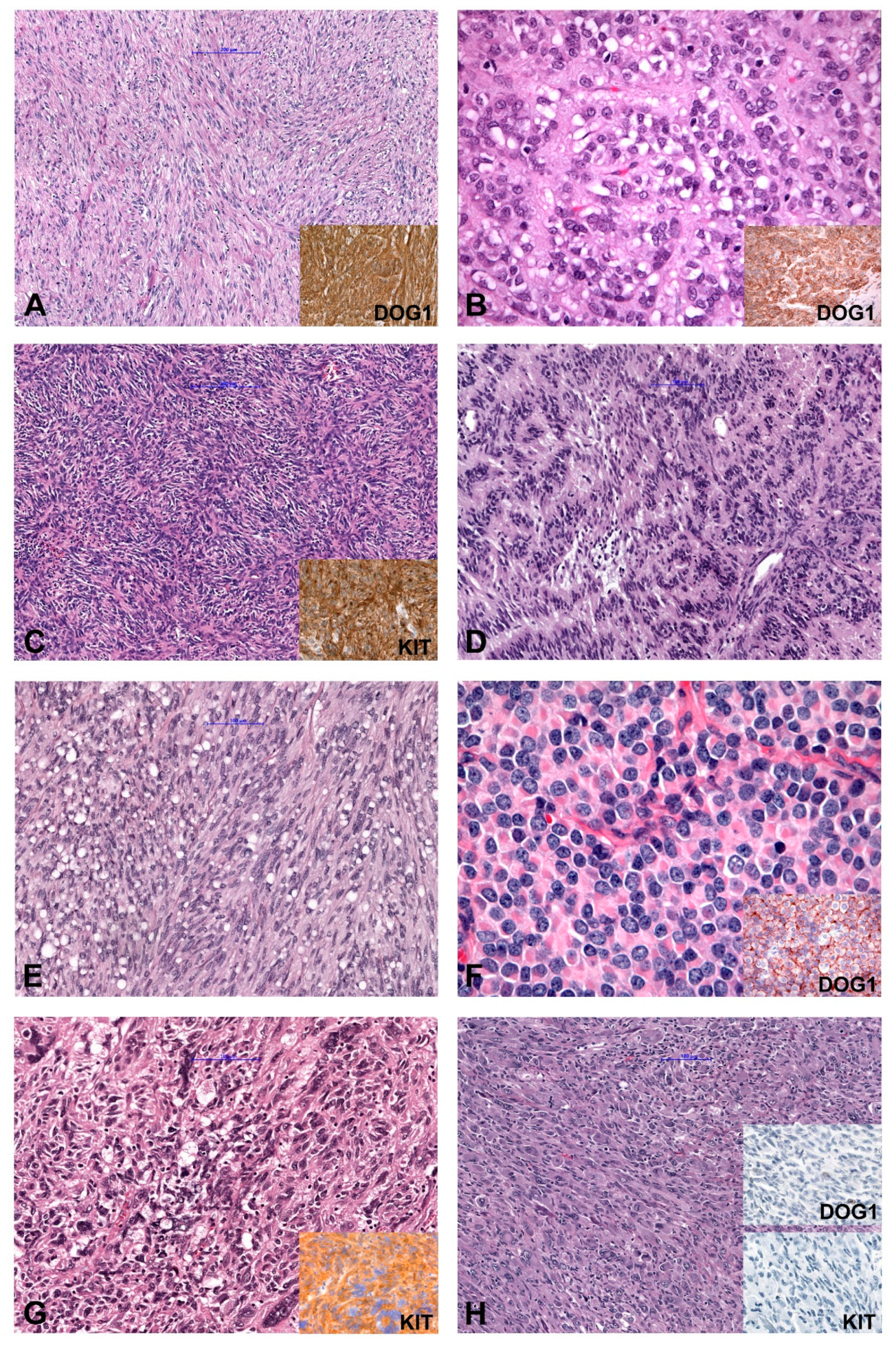
2. Molecular Classification
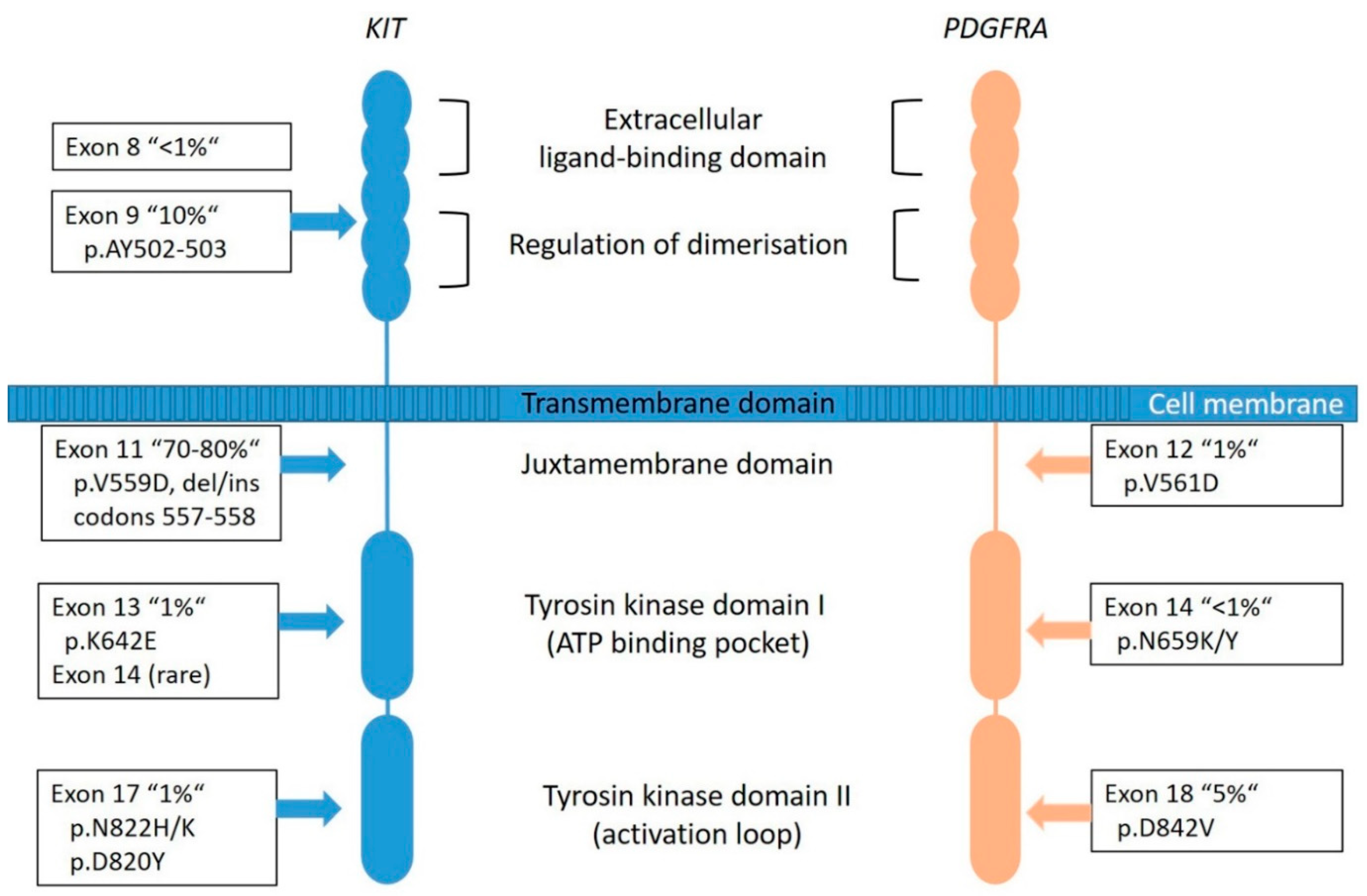
2.1. KIT/PDGFRA-Mutated GIST
2.1.1. KIT-Mutated GISTs
2.1.2. PDGFRA-Mutated GISTs
2.1.3. GIST Genomic Progression Model
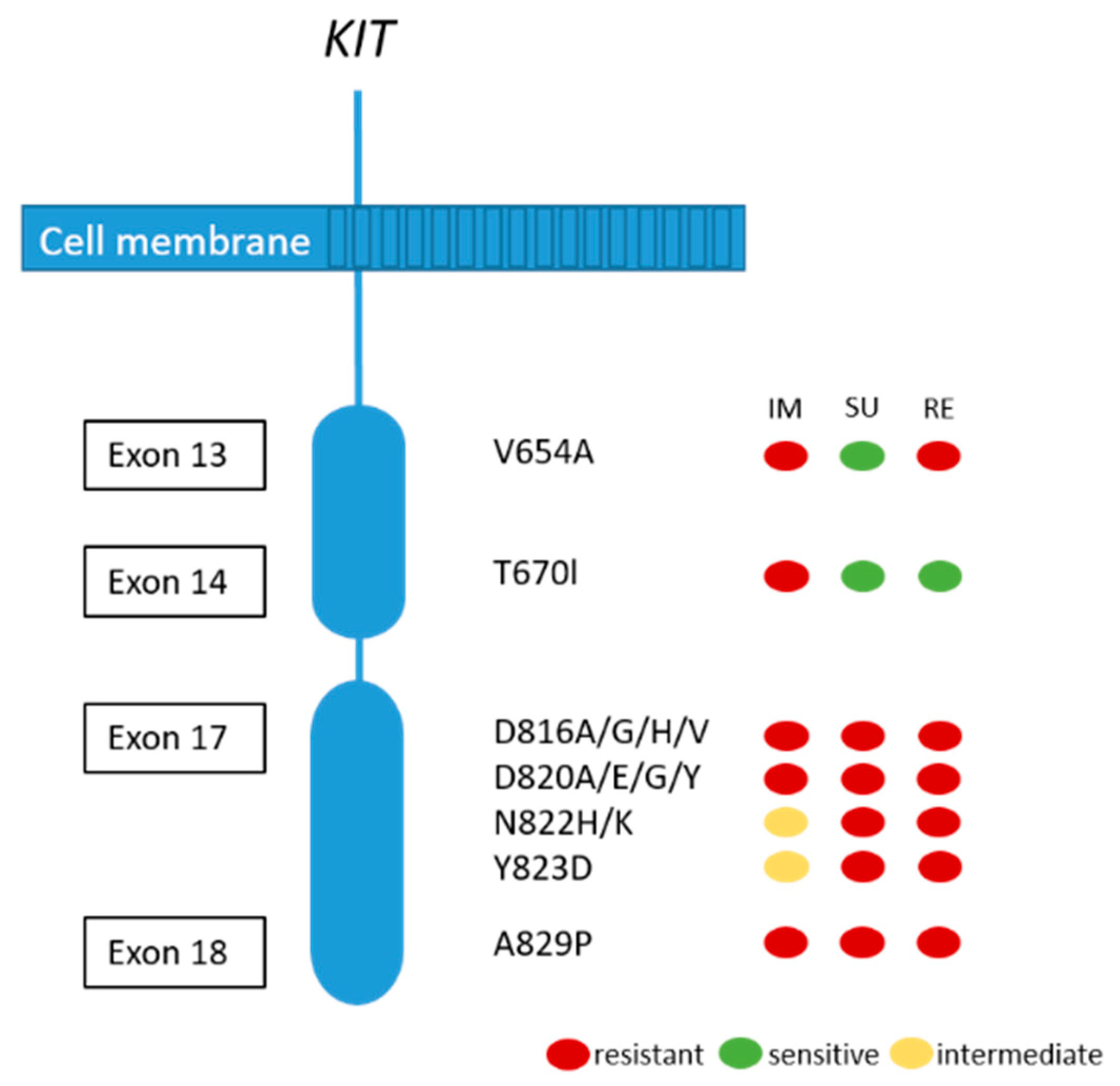
2.1.4. Resistance Mechanisms in GIST
2.1.5. Prognosis and Mutational Status in Treatment-Naïve GIST
2.1.6. Genetic Subtypes of GIST—Impact on Treatment Response
2.1.7. Morphological Changes after TKI Therapy
2.1.8. Familial GIST
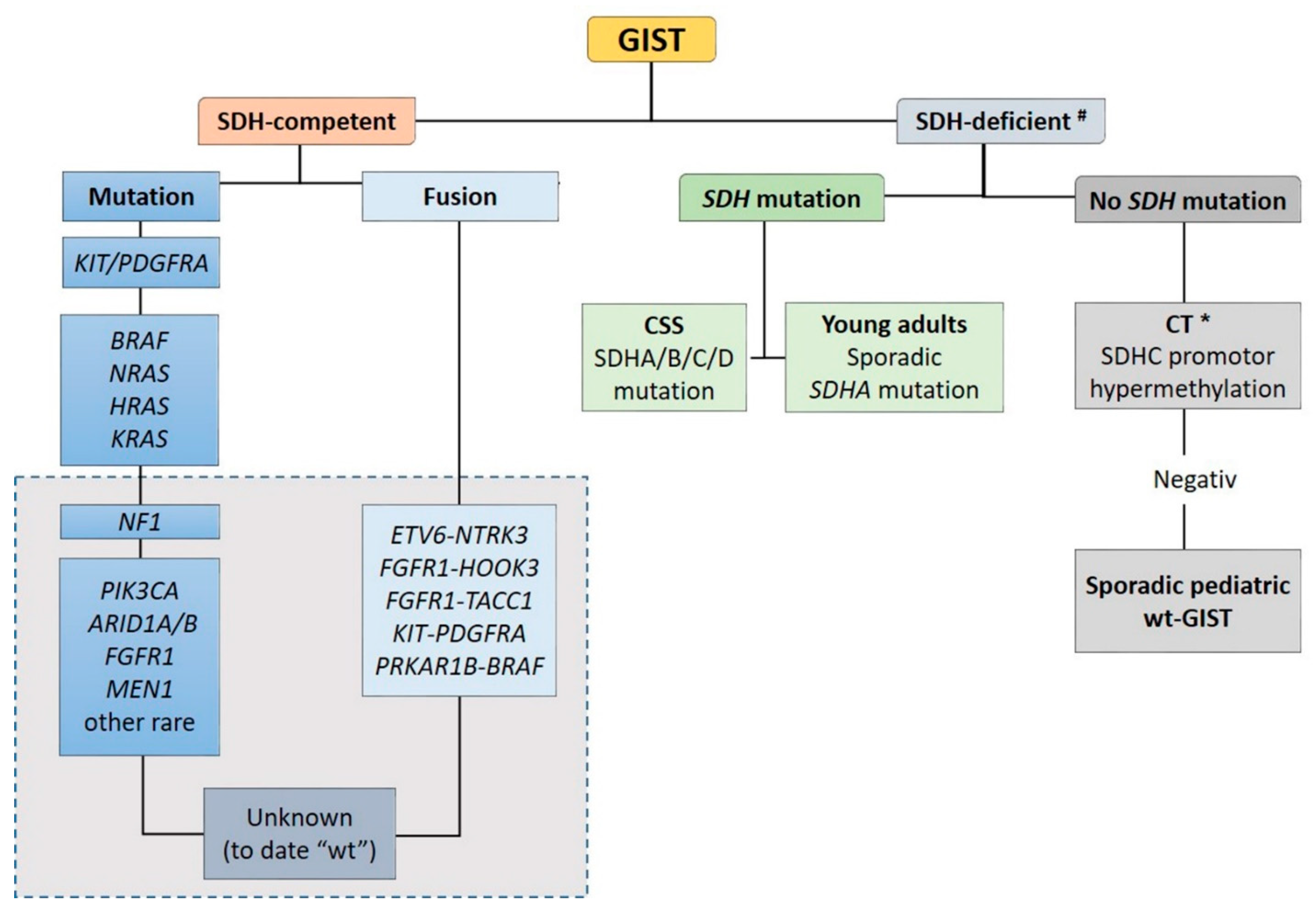
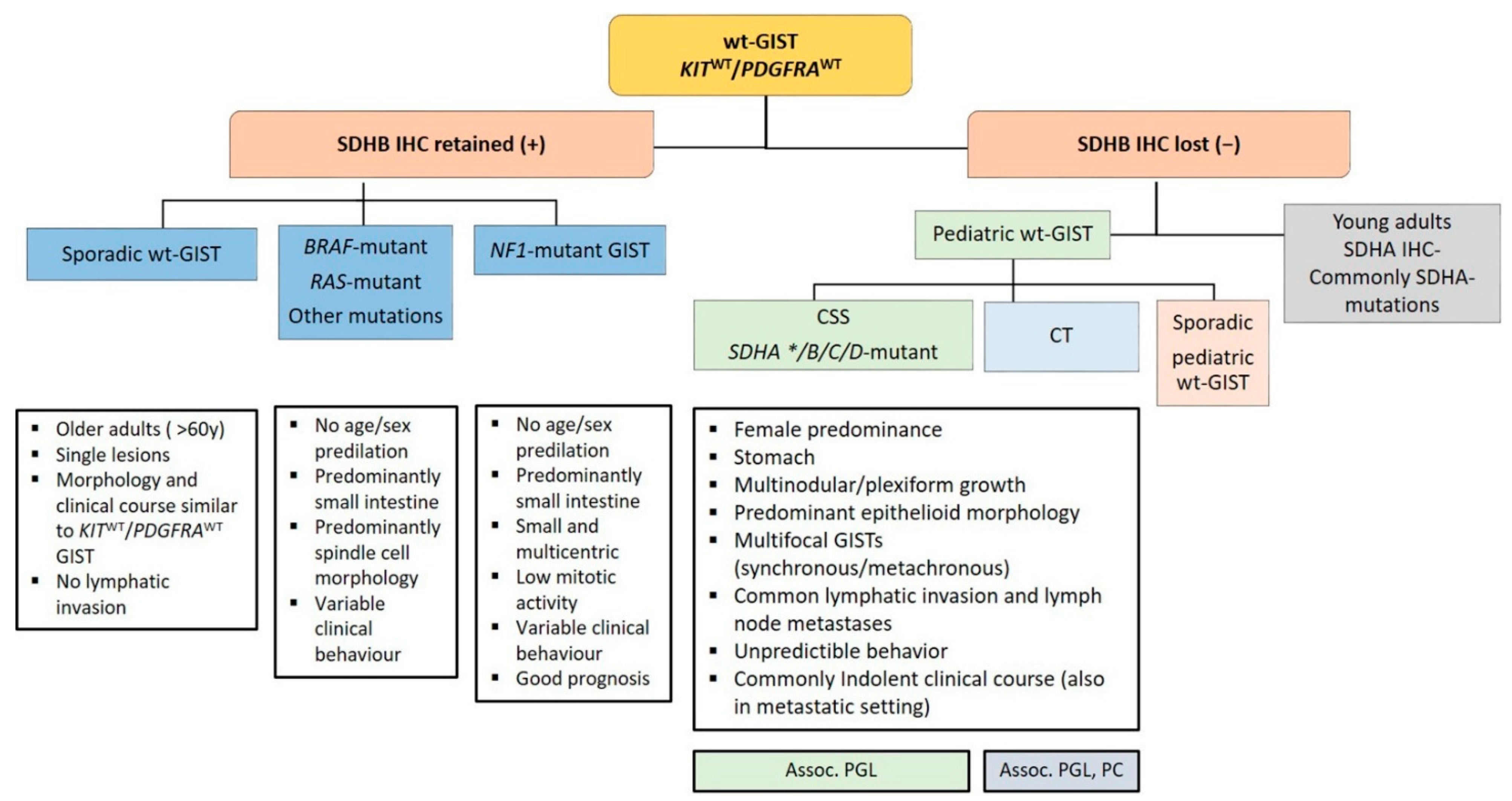
2.2. Wild-Type GIST
2.2.1. SDH-Competent wt-GISTs
NF1-Mutant GIST
BRAF, KRAS and PIK3CA-Mutant GISTs
GISTs with ETV6-NTRK3-Fusion
2.2.2. SDH-Deficient wt-GISTs
2.2.3. Syndromic SDH-Deficient wt-GIST
Carney Triad (CT)
Carney-Stratakis Syndrome (CSS)
SDHA-Deficient wt-GIST
2.2.4. Treatment Options in GISTs without Currently Druggable Target
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Soreide, K.; Sandvik, O.M.; Soreide, J.A.; Giljaca, V.; Jureckova, A.; Bulusu, V.R. Global epidemiology of gastrointestinal stromal tumours (gist): A systematic review of population-based cohort studies. Cancer Epidemiol. 2016, 40, 39–46. [Google Scholar] [CrossRef]
- Liegl-Atzwanger, B.; Fletcher, J.A.; Fletcher, C.D. Gastrointestinal stromal tumors. Virchows Arch. Int. J. Pathol. 2010, 456, 111–127. [Google Scholar] [CrossRef]
- Kindblom, L.G.; Remotti, H.E.; Aldenborg, F.; Meis-Kindblom, J.M. Gastrointestinal pacemaker cell tumor (gipact): Gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of cajal. Am. J. Pathol. 1998, 152, 1259–1269. [Google Scholar] [PubMed]
- Barajas-López, C.; Berezin, I.; Daniel, E.E.; Huizinga, J.D. Pacemaker activity recorded in interstitial cells of cajal of the gastrointestinal tract. Am. J. Physiol. 1989, 257, C830–C835. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors: Pathology and prognosis at different sites. Semin. Diagn. Pathol. 2006, 23, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Miceli, R.; Messerini, L.; Bearzi, I.; Mazzoleni, G.; Capella, C.; Arrigoni, G.; Sonzogni, A.; Sidoni, A.; Toffolatti, L.; et al. Natural history of imatinib-naive gists: A retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am. J. Surg. Pathol. 2011, 35, 1646–1656. [Google Scholar] [CrossRef]
- Miettinen, M.; Fetsch, J.F.; Sobin, L.H.; Lasota, J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: A clinicopathologic and molecular genetic study of 45 cases. Am. J. Surg. Pathol. 2006, 30, 90–96. [Google Scholar] [CrossRef]
- Nishida, T.; Hirota, S.; Taniguchi, M.; Hashimoto, K.; Isozaki, K.; Nakamura, H.; Kanakura, Y.; Tanaka, T.; Takabayashi, A.; Matsuda, H.; et al. Familial gastrointestinal stromal tumours with germline mutation of the kit gene. Nat. Genet. 1998, 19, 323–324. [Google Scholar] [CrossRef]
- Boikos, S.A.; Pappo, A.S.; Killian, J.K.; LaQuaglia, M.P.; Weldon, C.B.; George, S.; Trent, J.C.; von Mehren, M.; Wright, J.A.; Schiffman, J.D.; et al. Molecular subtypes of kit/pdgfra wild-type gastrointestinal stromal tumors: A report from the national institutes of health gastrointestinal stromal tumor clinic. JAMA Oncol. 2016, 2, 922–928. [Google Scholar] [CrossRef]
- Belinsky, M.G.; Rink, L.; von Mehren, M. Succinate dehydrogenase deficiency in pediatric and adult gastrointestinal stromal tumors. Front. Oncol. 2013, 3, 117. [Google Scholar] [CrossRef]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. Pdgfra activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the abl tyrosine kinase on the growth of bcr-abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Owzar, K.; Corless, C.L.; Hollis, D.; Borden, E.C.; Fletcher, C.D.; Ryan, C.W.; von Mehren, M.; Blanke, C.D.; Rankin, C.; et al. Correlation of kinase genotype and clinical outcome in the north american intergroup phase iii trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: Calgb 150105 study by cancer and leukemia group b and southwest oncology group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 5360–5367. [Google Scholar]
- Janeway, K.A.; Albritton, K.H.; Van Den Abbeele, A.D.; D’Amato, G.Z.; Pedrazzoli, P.; Siena, S.; Picus, J.; Butrynski, J.E.; Schlemmer, M.; Heinrich, M.C.; et al. Sunitinib treatment in pediatric patients with advanced gist following failure of imatinib. Pediatric Blood Cancer 2009, 52, 767–771. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schöffski, P.; Serrano, C.; Kang, Y.K.; Cassier, P.A.; Mir, O.; Eskens, F.; Tap, W.D.; et al. Avapritinib in advanced pdgfra d842v-mutant gastrointestinal stromal tumour (navigator): A multicentre, open-label, phase 1 trial. Lancet Oncol. 2020, 21, 935–946. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet (Lond. Engl.) 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (grid): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet (Lond. Engl.) 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Casali, P.G.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brodowicz, T.; et al. Gastrointestinal stromal tumours: Esmo-euracan clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2018, 29, iv68–iv78. [Google Scholar] [CrossRef]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors: Review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch. Pathol. Lab. Med. 2006, 130, 1466–1478. [Google Scholar] [PubMed]
- Fletcher, C.D.; Berman, J.J.; Corless, C.; Gorstein, F.; Lasota, J.; Longley, B.J.; Miettinen, M.; O’Leary, T.J.; Remotti, H.; Rubin, B.P.; et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Int. J. Surg. Pathol. 2002, 10, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Romeo, S.; Zhang, L.; Nafa, K.; Hornick, J.L.; Nielsen, G.P.; Mino-Kenudson, M.; Huang, H.Y.; Mosquera, J.M.; Dei Tos, P.A.; et al. Dedifferentiation in gastrointestinal stromal tumor to an anaplastic kit-negative phenotype: A diagnostic pitfall: Morphologic and molecular characterization of 8 cases occurring either de novo or after imatinib therapy. Am. J. Surg. Pathol. 2013, 37, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Díaz Delgado, M.; Hernández Amate, A.; Pereira Gallardo, S.; Jaramillo, S.; Virizuela Echaburu, J.A.; González-Cámpora, R.J. Gastrointestinal stromal tumors: Morphological, immunohistochemical and molecular changes associated with kinase inhibitor therapy. Pathol. Oncol. Res. POR 2011, 17, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Liegl, B.; Hornick, J.L.; Antonescu, C.R.; Corless, C.L.; Fletcher, C.D. Rhabdomyosarcomatous differentiation in gastrointestinal stromal tumors after tyrosine kinase inhibitor therapy: A novel form of tumor progression. Am. J. Surg. Pathol. 2009, 33, 218–226. [Google Scholar] [CrossRef]
- Choi, J.J.; Sinada-Bottros, L.; Maker, A.V.; Weisenberg, E. Dedifferentiated gastrointestinal stromal tumor arising de novo from the small intestine. Pathol. Res. Pract. 2014, 210, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, P.; Debiec-Rychter, M.; Stul, M.; De Wever, I.; Van Oosterom, A.T.; Sciot, R. Changing phenotype of gastrointestinal stromal tumours under imatinib mesylate treatment: A potential diagnostic pitfall. Histopathology 2005, 47, 41–47. [Google Scholar] [CrossRef]
- Sarlomo-Rikala, M.; Kovatich, A.J.; Barusevicius, A.; Miettinen, M. Cd117: A sensitive marker for gastrointestinal stromal tumors that is more specific than cd34. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 1998, 11, 728–734. [Google Scholar]
- Liegl, B.; Hornick, J.L.; Corless, C.L.; Fletcher, C.D. Monoclonal antibody dog1.1 shows higher sensitivity than kit in the diagnosis of gastrointestinal stromal tumors, including unusual subtypes. Am. J. Surg. Pathol. 2009, 33, 437–446. [Google Scholar] [CrossRef]
- Miettinen, M.; Wang, Z.F.; Lasota, J. Dog1 antibody in the differential diagnosis of gastrointestinal stromal tumors: A study of 1840 cases. Am. J. Surg. Pathol. 2009, 33, 1401–1408. [Google Scholar] [CrossRef]
- Espinosa, I.; Lee, C.H.; Kim, M.K.; Rouse, B.T.; Subramanian, S.; Montgomery, K.; Varma, S.; Corless, C.L.; Heinrich, M.C.; Smith, K.S.; et al. A novel monoclonal antibody against dog1 is a sensitive and specific marker for gastrointestinal stromal tumors. Am. J. Surg. Pathol. 2008, 32, 210–218. [Google Scholar] [CrossRef]
- Lopes, L.F.; West, R.B.; Bacchi, L.M.; van de Rijn, M.; Bacchi, C.E. Dog1 for the diagnosis of gastrointestinal stromal tumor (gist): Comparison between 2 different antibodies. Appl. Immunohistochem. Mol. Morphol. AIMM 2010, 18, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.P.; Singer, S.; Tsao, C.; Duensing, A.; Lux, M.L.; Ruiz, R.; Hibbard, M.K.; Chen, C.J.; Xiao, S.; Tuveson, D.A.; et al. Kit activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001, 61, 8118–8121. [Google Scholar] [PubMed]
- Wozniak, A.; Rutkowski, P.; Piskorz, A.; Ciwoniuk, M.; Osuch, C.; Bylina, E.; Sygut, J.; Chosia, M.; Rys, J.; Urbanczyk, K.; et al. Prognostic value of kit/pdgfra mutations in gastrointestinal stromal tumours (gist): Polish clinical gist registry experience. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2012, 23, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.P.; Wong, G.C.; Guo, T.; Maki, R.G.; Singer, S.; Dematteo, R.P.; Besmer, P.; Antonescu, C.R. Novel v600e braf mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 2008, 47, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Janeway, K.A.; Liegl, B.; Harlow, A.; Le, C.; Perez-Atayde, A.; Kozakewich, H.; Corless, C.L.; Heinrich, M.C.; Fletcher, J.A. Pediatric kit wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share kit activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res. 2007, 67, 9084–9088. [Google Scholar] [CrossRef] [PubMed]
- Shi, E.; Chmielecki, J.; Tang, C.M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. Fgfr1 and ntrk3 actionable alterations in “wild-type” gastrointestinal stromal tumors. J. Transl. Med. 2016, 14, 339. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, M.A.; Astolfi, A.; Di Battista, M.; Heinrich, M.C.; Paterini, P.; Scotlandi, K.; Santini, D.; Catena, F.; Manara, M.C.; Nannini, M.; et al. Insulin-like growth factor 1 receptor expression in wild-type gists: A potential novel therapeutic target. Int. J. Cancer 2009, 125, 2991–2994. [Google Scholar] [CrossRef]
- Charo, L.M.; Burgoyne, A.M.; Fanta, P.T.; Patel, H.; Chmielecki, J.; Sicklick, J.K.; McHale, M.T. A novel prkar1b-braf fusion in gastrointestinal stromal tumor guides adjuvant treatment decision-making during pregnancy. J. Natl. Compr. Cancer Netw. JNCCN 2018, 16, 238–242. [Google Scholar] [CrossRef]
- Brenca, M.; Rossi, S.; Polano, M.; Gasparotto, D.; Zanatta, L.; Racanelli, D.; Valori, L.; Lamon, S.; Dei Tos, A.P.; Maestro, R. Transcriptome sequencing identifies etv6-ntrk3 as a gene fusion involved in gist. J. Pathol. 2016, 238, 543–549. [Google Scholar] [CrossRef]
- Hostein, I.; Faur, N.; Primois, C.; Boury, F.; Denard, J.; Emile, J.F.; Bringuier, P.P.; Scoazec, J.Y.; Coindre, J.M. Braf mutation status in gastrointestinal stromal tumors. Am. J. Clin. Pathol. 2010, 133, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Remillard, S.P.; Zhang, Y.X.; Doyle, L.A.; George, S.; Hornick, J.L. Loss of expression of sdha predicts sdha mutations in gastrointestinal stromal tumors. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2013, 26, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour Babaei, M.; Kamalidehghan, B.; Saleem, M.; Huri, H.Z.; Ahmadipour, F. Receptor tyrosine kinase (c-kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Dev. Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Yamagata, A.; Nishikawa, S.; Yoshinaga, K.; Kobayashi, S.; Nishi, K.; Nishikawa, S. Requirement of c-kit for development of intestinal pacemaker system. Development (Camb. Engl.) 1992, 116, 369–375. [Google Scholar]
- Orr-Urtreger, A.; Avivi, A.; Zimmer, Y.; Givol, D.; Yarden, Y.; Lonai, P. Developmental expression of c-kit, a proto-oncogene encoded by the w locus. Development (Camb. Engl.) 1990, 109, 911–923. [Google Scholar]
- Heinrich, M.C.; Rubin, B.P.; Longley, B.J.; Fletcher, J.A. Biology and genetic aspects of gastrointestinal stromal tumors: Kit activation and cytogenetic alterations. Hum. Pathol. 2002, 33, 484–495. [Google Scholar] [CrossRef]
- Wang, W.J.; Li, H.T.; Yu, J.P.; Li, Y.M.; Han, X.P.; Chen, P.; Yu, W.W.; Chen, W.K.; Jiao, Z.Y.; Liu, H.B. Identification of key genes and associated pathways in kit/pdgfra wild-type gastrointestinal stromal tumors through bioinformatics analysis. Mol. Med. Rep. 2018, 18, 4499–4515. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Sommer, G.; Sarran, L.; Tschernyavsky, S.J.; Riedel, E.; Woodruff, J.M.; Robson, M.; Maki, R.; Brennan, M.F.; Ladanyi, M.; et al. Association of kit exon 9 mutations with nongastric primary site and aggressive behavior: Kit mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 3329–3337. [Google Scholar]
- Wozniak, A.; Rutkowski, P.; Schöffski, P.; Ray-Coquard, I.; Hostein, I.; Schildhaus, H.U.; Le Cesne, A.; Bylina, E.; Limon, J.; Blay, J.Y.; et al. Tumor genotype is an independent prognostic factor in primary gastrointestinal stromal tumors of gastric origin: A european multicenter analysis based on conticagist. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6105–6116. [Google Scholar] [CrossRef]
- Debiec-Rychter, M.; Sciot, R.; Le Cesne, A.; Schlemmer, M.; Hohenberger, P.; van Oosterom, A.T.; Blay, J.Y.; Leyvraz, S.; Stul, M.; Casali, P.G.; et al. Kit mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur. J. Cancer (Oxf. Engl. 1990) 2006, 42, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Steigen, S.E.; Eide, T.J.; Wasag, B.; Lasota, J.; Miettinen, M. Mutations in gastrointestinal stromal tumors--a population-based study from northern norway. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2007, 115, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.Y.; Ma, X.L.; Wang, M.; Zhuang, C.; Ni, B. Exon 11 homozygous mutations and intron 10/exon 11 junction deletions in the kit gene are associated with poor prognosis of patients with gastrointestinal stromal tumors. Cancer Med. 2020, 9, 6485–6496. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Mariño-Enríquez, A.; Fletcher, J.A. What is new in gastrointestinal stromal tumor? Adv. Anat. Pathol. 2017, 24, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Lasota, J.; Corless, C.L.; Heinrich, M.C.; Debiec-Rychter, M.; Sciot, R.; Wardelmann, E.; Merkelbach-Bruse, S.; Schildhaus, H.U.; Steigen, S.E.; Stachura, J.; et al. Clinicopathologic profile of gastrointestinal stromal tumors (gists) with primary kit exon 13 or exon 17 mutations: A multicenter study on 54 cases. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2008, 21, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Szucs, Z.; Thway, K.; Fisher, C.; Bulusu, R.; Constantinidou, A.; Benson, C.; van der Graaf, W.T.; Jones, R.L. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol. (Lond. Engl.) 2017, 13, 93–107. [Google Scholar] [CrossRef]
- Fredriksson, L.; Li, H.; Eriksson, U. The pdgf family: Four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004, 15, 197–204. [Google Scholar] [CrossRef]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. Pdgfra mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef]
- Breiner, J.A.; Meis-Kindblom, J.; Kindblom, L.G.; McComb, E.; Liu, J.; Nelson, M.; Bridge, J.A. Loss of 14q and 22q in gastrointestinal stromal tumors (pacemaker cell tumors). Cancer Genet. Cytogenet. 2000, 120, 111–116. [Google Scholar] [CrossRef]
- Belinsky, M.G.; Rink, L.; Cai, K.Q.; Capuzzi, S.J.; Hoang, Y.; Chien, J.; Godwin, A.K.; von Mehren, M. Somatic loss of function mutations in neurofibromin 1 and myc associated factor x genes identified by exome-wide sequencing in a wild-type gist case. BMC Cancer 2015, 15, 887. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Wang, Y.; Liang, C.W.; Bahri, N.; Quattrone, A.; Doyle, L.; Mariño-Enríquez, A.; Lauria, A.; Zhu, M.; Debiec-Rychter, M.; et al. Max inactivation is an early event in gist development that regulates p16 and cell proliferation. Nat. Commun. 2017, 8, 14674. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.; Sciot, R.; Guillou, L.; Pauwels, P.; Wasag, B.; Stul, M.; Vermeesch, J.R.; Vandenberghe, P.; Limon, J.; Debiec-Rychter, M. Array cgh analysis in primary gastrointestinal stromal tumors: Cytogenetic profile correlates with anatomic site and tumor aggressiveness, irrespective of mutational status. Genes Chromosomes Cancer 2007, 46, 261–276. [Google Scholar] [CrossRef] [PubMed]
- El-Rifai, W.; Sarlomo-Rikala, M.; Andersson, L.C.; Knuutila, S.; Miettinen, M. DNA sequence copy number changes in gastrointestinal stromal tumors: Tumor progression and prognostic significance. Cancer Res. 2000, 60, 3899–3903. [Google Scholar] [PubMed]
- Gunawan, B.; Bergmann, F.; Höer, J.; Langer, C.; Schumpelick, V.; Becker, H.; Füzesi, L. Biological and clinical significance of cytogenetic abnormalities in low-risk and high-risk gastrointestinal stromal tumors. Hum. Pathol. 2002, 33, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, B.; von Heydebreck, A.; Sander, B.; Schulten, H.J.; Haller, F.; Langer, C.; Armbrust, T.; Bollmann, M.; Gasparov, S.; Kovac, D.; et al. An oncogenetic tree model in gastrointestinal stromal tumours (gists) identifies different pathways of cytogenetic evolution with prognostic implications. J. Pathol. 2007, 211, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Delfs, C.; Cameron, S.; Gunawan, B.; Agaimy, A.; Ghadimi, B.M.; Haller, F. Chromosomal aberrations in primary pdgfra-mutated gastrointestinal stromal tumors. Hum. Pathol. 2014, 45, 85–97. [Google Scholar] [CrossRef]
- Schneider-Stock, R.; Boltze, C.; Lasota, J.; Miettinen, M.; Peters, B.; Pross, M.; Roessner, A.; Günther, T. High prognostic value of p16ink4 alterations in gastrointestinal stromal tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 1688–1697. [Google Scholar] [CrossRef]
- Merten, L.; Agaimy, A.; Moskalev, E.A.; Giedl, J.; Kayser, C.; Geddert, H.; Schaefer, I.M.; Cameron, S.; Werner, M.; Ströbel, P.; et al. Inactivating mutations of rb1 and tp53 correlate with sarcomatous histomorphology and metastasis/recurrence in gastrointestinal stromal tumors. Am. J. Clin. Pathol. 2016, 146, 718–726. [Google Scholar] [CrossRef]
- Romeo, S.; Debiec-Rychter, M.; Van Glabbeke, M.; Van Paassen, H.; Comite, P.; Van Eijk, R.; Oosting, J.; Verweij, J.; Terrier, P.; Schneider, U.; et al. Cell cycle/apoptosis molecule expression correlates with imatinib response in patients with advanced gastrointestinal stromal tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 4191–4198. [Google Scholar] [CrossRef]
- Wang, Y.; Marino-Enriquez, A.; Bennett, R.R.; Zhu, M.; Shen, Y.; Eilers, G.; Lee, J.C.; Henze, J.; Fletcher, B.S.; Gu, Z.; et al. Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat. Genet. 2014, 46, 601–606. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Structure and regulation of kit protein-tyrosine kinase--the stem cell factor receptor. Biochem. Biophys. Res. Commun. 2005, 338, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Liegl, B.; Kepten, I.; Le, C.; Zhu, M.; Demetri, G.D.; Heinrich, M.C.; Fletcher, C.D.; Corless, C.L.; Fletcher, J.A. Heterogeneity of kinase inhibitor resistance mechanisms in gist. J. Pathol. 2008, 216, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Blanke, C.D.; Demetri, G.D.; Joensuu, H.; Roberts, P.J.; Eisenberg, B.L.; von Mehren, M.; Fletcher, C.D.; Sandau, K.; et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 4764–4774. [Google Scholar] [CrossRef]
- Agaram, N.P.; Besmer, P.; Wong, G.C.; Guo, T.; Socci, N.D.; Maki, R.G.; DeSantis, D.; Brennan, M.F.; Singer, S.; DeMatteo, R.P.; et al. Pathologic and molecular heterogeneity in imatinib-stable or imatinib-responsive gastrointestinal stromal tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Prenen, H.; Cools, J.; Mentens, N.; Folens, C.; Sciot, R.; Schöffski, P.; Van Oosterom, A.; Marynen, P.; Debiec-Rychter, M. Efficacy of the kinase inhibitor su11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 2622–2627. [Google Scholar] [CrossRef]
- Wardelmann, E.; Merkelbach-Bruse, S.; Pauls, K.; Thomas, N.; Schildhaus, H.U.; Heinicke, T.; Speidel, N.; Pietsch, T.; Buettner, R.; Pink, D.; et al. Polyclonal evolution of multiple secondary kit mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1743–1749. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Maki, R.G.; Corless, C.L.; Antonescu, C.R.; Harlow, A.; Griffith, D.; Town, A.; McKinley, A.; Ou, W.B.; Fletcher, J.A.; et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 5352–5359. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Bosbach, B.; Loo, J.K.; Vitiello, G.A. The v654a second-site kit mutation increases tumor oncogenesis and stat activation in a mouse model of gastrointestinal stromal tumor. Oncogene 2020, 39, 7153–7165. [Google Scholar] [CrossRef]
- Dalle Fratte, C.; Guardascione, M.; De Mattia, E.; Borsatti, E.; Boschetto, R.; Farruggio, A.; Canzonieri, V.; Romanato, L.; Borsatti, R.; Gagno, S.; et al. Clonal selection of a novel deleterious tp53 somatic mutation discovered in ctdna of a kit/pdgfra wild-type gastrointestinal stromal tumor resistant to imatinib. Front. Pharmacol. 2020, 11, 36. [Google Scholar] [CrossRef]
- Serrano, C.; Fletcher, J.A. Overcoming heterogenity in imatinib-resistant gastrointestinal stromal tumor. Oncotarget 2019, 10, 6286–6287. [Google Scholar] [CrossRef]
- Grunewald, S.; Klug, L.R.; Muhlenberg, T. Resistance to avapritinib in pdgfra-driven gist is caused by secondary mutations in the pdgfra kinase domain. Cancer Discov. 2021, 11, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Roberts, P.J.; Sarlomo-Rikala, M.; Andersson, L.C.; Tervahartiala, P.; Tuveson, D.; Silberman, S.; Capdeville, R.; Dimitrijevic, S.; Druker, B.; et al. Effect of the tyrosine kinase inhibitor sti571 in a patient with a metastatic gastrointestinal stromal tumor. N. Engl. J. Med. 2001, 344, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.Y.; Serrano, C.; Heinrich, M.C.; Zalcberg, J.; Bauer, S.; Gelderblom, H.; Schöffski, P.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (invictus): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 923–934. [Google Scholar] [CrossRef]
- Wardelmann, E.; Losen, I.; Hans, V.; Neidt, I.; Speidel, N.; Bierhoff, E.; Heinicke, T.; Pietsch, T.; Büttner, R.; Merkelbach-Bruse, S. Deletion of trp-557 and lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int. J. Cancer 2003, 106, 887–895. [Google Scholar] [CrossRef]
- Rutkowski, P.; Magnan, H.; Chou, A.J.; Benson, C. Treatment of gastrointestinal stromal tumours in paediatric and young adult patients with sunitinib: A multicentre case series. BMC Cancer 2017, 17, 717. [Google Scholar] [CrossRef]
- Li, L.; Khalili, M. Case report of rhabdomyosarcomatous transformation of a primary gastrointestinal stromal tumor (gist). BMC Cancer 2019, 19, 913. [Google Scholar] [CrossRef]
- Chompret, A.; Kannengiesser, C.; Barrois, M.; Terrier, P.; Dahan, P.; Tursz, T.; Lenoir, G.M.; Bressac-De Paillerets, B. Pdgfra germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 2004, 126, 318–321. [Google Scholar] [CrossRef]
- Hirota, S.; Nishida, T.; Isozaki, K.; Taniguchi, M.; Nishikawa, K.; Ohashi, A.; Takabayashi, A.; Obayashi, T.; Okuno, T.; Kinoshita, K.; et al. Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of kit gene. Gastroenterology 2002, 122, 1493–1499. [Google Scholar] [CrossRef]
- Beghini, A.; Tibiletti, M.G.; Roversi, G.; Chiaravalli, A.M.; Serio, G.; Capella, C.; Larizza, L. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer 2001, 92, 657–662. [Google Scholar] [CrossRef]
- Lasota, J.; Miettinen, M. A new familial gist identified. Am. J. Surg. Pathol. 2006, 30, 1342. [Google Scholar] [CrossRef]
- Robson, M.E.; Glogowski, E.; Sommer, G.; Antonescu, C.R.; Nafa, K.; Maki, R.G.; Ellis, N.; Besmer, P.; Brennan, M.; Offit, K. Pleomorphic characteristics of a germ-line kit mutation in a large kindred with gastrointestinal stromal tumors, hyperpigmentation, and dysphagia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 1250–1254. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Wardelmann, E.; Ma, Y.; Merkelbach-Bruse, S.; Preussner, L.M.; Woolery, C.; Baldus, S.E.; Heinicke, T.; Thiele, J.; Buettner, R.; et al. Novel germline mutation of kit associated with familial gastrointestinal stromal tumors and mastocytosis. Gastroenterology 2005, 129, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Arima, J.; Hiramatsu, M.; Taniguchi, K.; Kobayashi, T.; Tsunematsu, I.; Kagota, S.; Sakane, J.; Suzuki, Y.; Hirota, S. Multiple gastrointestinal stromal tumors caused by a novel germline kit gene mutation (asp820gly): A case report and literature review. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2020, 23, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Fornasarig, M.; Gasparotto, D.; Foltran, L.; Campigotto, M. A novel kindred with familial gastrointestinal stromal tumors caused by a rare kit germline mutation (n655k): Clinico-pathological presentation and tki sensitivity. J. Pers. Med. 2020, 10, 234. [Google Scholar] [CrossRef]
- Kang, D.Y.; Park, C.K.; Choi, J.S.; Jin, S.Y.; Kim, H.J.; Joo, M.; Kang, M.S.; Moon, W.S.; Yun, K.J.; Yu, E.S.; et al. Multiple gastrointestinal stromal tumors: Clinicopathologic and genetic analysis of 12 patients. Am. J. Surg. Pathol. 2007, 31, 224–232. [Google Scholar] [CrossRef]
- Neuhann, T.M.; Mansmann, V.; Merkelbach-Bruse, S.; Klink, B.; Hellinger, A.; Höffkes, H.G.; Wardelmann, E.; Schildhaus, H.U.; Tinschert, S. A novel germline kit mutation (p.L576p) in a family presenting with juvenile onset of multiple gastrointestinal stromal tumors, skin hyperpigmentations, and esophageal stenosis. Am. J. Surg. Pathol. 2013, 37, 898–905. [Google Scholar] [CrossRef]
- Bachet, J.B.; Landi, B.; Laurent-Puig, P.; Italiano, A.; Le Cesne, A.; Lévy, P.; Safar, V.; Duffaud, F.; Blay, J.Y.; Emile, J.F. Diagnosis, prognosis and treatment of patients with gastrointestinal stromal tumour (gist) and germline mutation of kit exon 13. Eur. J. Cancer (Oxf. Engl. 1990) 2013, 49, 2531–2541. [Google Scholar] [CrossRef]
- Graham, J.; Debiec-Rychter, M.; Corless, C.L.; Reid, R.; Davidson, R.; White, J.D. Imatinib in the management of multiple gastrointestinal stromal tumors associated with a germline kit k642e mutation. Arch. Pathol. Lab. Med. 2007, 131, 1393–1396. [Google Scholar]
- Thalheimer, A.; Schlemmer, M.; Bueter, M.; Merkelbach-Bruse, S.; Schildhaus, H.U.; Buettner, R.; Hartung, E.; Thiede, A.; Meyer, D.; Fein, M.; et al. Familial gastrointestinal stromal tumors caused by the novel kit exon 17 germline mutation n822y. Am. J. Surg. Pathol. 2008, 32, 1560–1565. [Google Scholar] [CrossRef]
- Pasini, B.; Matyakhina, L.; Bei, T.; Muchow, M.; Boikos, S.; Ferrando, B.; Carney, J.A.; Stratakis, C.A. Multiple gastrointestinal stromal and other tumors caused by platelet-derived growth factor receptor alpha gene mutations: A case associated with a germline v561d defect. J. Clin. Endocrinol. Metab. 2007, 92, 3728–3732. [Google Scholar] [CrossRef]
- Manley, P.N.; Abu-Abed, S.; Kirsch, R.; Hawrysh, A.; Perrier, N.; Feilotter, H.; Pollett, A.; Riddell, R.H.; Hookey, L.; Walia, J.S. Familial pdgfra-mutation syndrome: Somatic and gastrointestinal phenotype. Hum. Pathol. 2018, 76, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hirota, S.; Isozaki, K.; Sun, H.; Ohashi, A.; Kinoshita, K.; O’Brien, P.; Kapusta, L.; Dardick, I.; Obayashi, T.; et al. Polyclonal nature of diffuse proliferation of interstitial cells of cajal in patients with familial and multiple gastrointestinal stromal tumours. Gut 2002, 51, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Wang, Z.F.; Sarlomo-Rikala, M.; Osuch, C.; Rutkowski, P.; Lasota, J. Succinate dehydrogenase-deficient gists: A clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric gists with predilection to young age. Am. J. Surg. Pathol. 2011, 35, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, D.; Rossi, S.; Polano, M.; Tamborini, E.; Lorenzetto, E.; Sbaraglia, M.; Mondello, A.; Massani, M.; Lamon, S.; Bracci, R.; et al. Quadruple-negative gist is a sentinel for unrecognized neurofibromatosis type 1 syndrome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, A.M.; De Siena, M.; Alkhuziem, M.; Tang, C.M.; Medina, B.; Fanta, P.T.; Belinsky, M.G.; von Mehren, M.; Thorson, J.A.; Madlensky, L.; et al. Duodenal-jejunal flexure gi stromal tumor frequently heralds somatic nf1 and notch pathway mutations. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Gaal, J.; Stratakis, C.A.; Carney, J.A.; Ball, E.R.; Korpershoek, E.; Lodish, M.B.; Levy, I.; Xekouki, P.; van Nederveen, F.H.; den Bakker, M.A.; et al. Sdhb immunohistochemistry: A useful tool in the diagnosis of carney-stratakis and carney triad gastrointestinal stromal tumors. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2011, 24, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Tobo, T.; Nakamori, M.; Imamura, M.; Kojima, A.; Oda, Y.; Nakamura, N.; Takahira, T.; Yao, T.; Tsuneyoshi, M. Neurofibromatosis type 1-related gastrointestinal stromal tumors: A special reference to loss of heterozygosity at 14q and 22q. J. Cancer Res. Clin. Oncol. 2009, 135, 791–798. [Google Scholar] [CrossRef]
- Agaimy, A.; Terracciano, L.M.; Dirnhofer, S.; Tornillo, L.; Foerster, A.; Hartmann, A.; Bihl, M.P. V600e braf mutations are alternative early molecular events in a subset of kit/pdgfra wild-type gastrointestinal stromal tumours. J. Clin. Pathol. 2009, 62, 613–616. [Google Scholar] [CrossRef]
- Daniels, M.; Lurkin, I.; Pauli, R.; Erbstösser, E.; Hildebrandt, U.; Hellwig, K.; Zschille, U.; Lüders, P.; Krüger, G.; Knolle, J.; et al. Spectrum of kit/pdgfra/braf mutations and phosphatidylinositol-3-kinase pathway gene alterations in gastrointestinal stromal tumors (gist). Cancer Lett. 2011, 312, 43–54. [Google Scholar] [CrossRef]
- Nannini, M.; Biasco, G.; Astolfi, A.; Pantaleo, M.A. An overview on molecular biology of kit/pdgfra wild type (wt) gastrointestinal stromal tumours (gist). J. Med Genet. 2013, 50, 653–661. [Google Scholar] [CrossRef]
- Falchook, G.S.; Trent, J.C.; Heinrich, M.C.; Beadling, C.; Patterson, J.; Bastida, C.C.; Blackman, S.C.; Kurzrock, R. Braf mutant gastrointestinal stromal tumor: First report of regression with braf inhibitor dabrafenib (gsk2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget 2013, 4, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.; Nucifora, M.; Molinari, F.; Conca, E.; Anania, M.C.; Bordoni, A.; Saletti, P.; Mazzucchelli, L.; Pilotti, S.; Pierotti, M.A.; et al. Kras and braf mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Lasota, J.; Felisiak-Golabek, A.; Wasag, B.; Kowalik, A.; Zięba, S.; Chłopek, M.; Wang, Z.F.; Coates, T.; Kopczynski, J.; Gozdz, S.; et al. Frequency and clinicopathologic profile of pik3ca mutant gists: Molecular genetic study of 529 cases. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2016, 29, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of larotrectinib in trk fusion-positive cancers in adults and children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic ntrk fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with trk fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. N. Engl. J. Med. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Marchiò, C.; Scaltriti, M.; Ladanyi, M.; Iafrate, A.J.; Bibeau, F.; Dietel, M.; Hechtman, J.F.; Troiani, T.; López-Rios, F.; Douillard, J.Y.; et al. Esmo recommendations on the standard methods to detect ntrk fusions in daily practice and clinical research. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2019, 30, 1417–1427. [Google Scholar] [CrossRef]
- Demetri, G.D.; Antonescu, C.R.; Bjerkehagen, B.; Bovée, J.; Boye, K.; Chacón, M.; Dei Tos, A.P.; Desai, J.; Fletcher, J.A.; Gelderblom, H.; et al. Diagnosis and management of tropomyosin receptor kinase (trk) fusion sarcomas: Expert recommendations from the world sarcoma network. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2020, 31, 1506–1517. [Google Scholar] [CrossRef]
- Brčić, I.; Godschachner, T.M.; Bergovec, M.; Igrec, J.; Till, H.; Lackner, H.; Scheipl, S.; Kashofer, K.; Brodowicz, T.; Leithner, A.; et al. Broadening the spectrum of ntrk rearranged mesenchymal tumors and usefulness of pan-trk immunohistochemistry for identification of ntrk fusions. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2021, 34, 396–407. [Google Scholar] [CrossRef]
- Mason, E.F.; Hornick, J.L. Succinate dehydrogenase deficiency is associated with decreased 5-hydroxymethylcytosine production in gastrointestinal stromal tumors: Implications for mechanisms of tumorigenesis. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2013, 26, 1492–1497. [Google Scholar] [CrossRef]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I., Jr.; Jahromi, M.S.; Xekouki, P.; et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J. Succinate dehydrogenase (sdh)-deficient neoplasia. Histopathology 2018, 72, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J. Succinate dehydrogenase (sdh) and mitochondrial driven neoplasia. Pathology 2012, 44, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, M.G.; Rink, L.; Cai, K.Q.; Ochs, M.F.; Eisenberg, B.; Huang, M.; von Mehren, M.; Godwin, A.K. The insulin-like growth factor system as a potential therapeutic target in gastrointestinal stromal tumors. Cell Cycle (Georget. Tex.) 2008, 7, 2949–2955. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chou, A.; Chen, J.; Clarkson, A.; Samra, J.S.; Clifton-Bligh, R.J.; Hugh, T.J.; Gill, A.J. Succinate dehydrogenase-deficient gists are characterized by igf1r overexpression. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2012, 25, 1307–1313. [Google Scholar] [CrossRef]
- Lasota, J.; Wang, Z.; Kim, S.Y.; Helman, L.; Miettinen, M. Expression of the receptor for type i insulin-like growth factor (igf1r) in gastrointestinal stromal tumors: An immunohistochemical study of 1078 cases with diagnostic and therapeutic implications. Am. J. Surg. Pathol. 2013, 37, 114–119. [Google Scholar] [CrossRef]
- Yu, H.; Rohan, T. Role of the insulin-like growth factor family in cancer development and progression. J. Natl. Cancer Inst. 2000, 92, 1472–1489. [Google Scholar] [CrossRef]
- Tarn, C.; Rink, L.; Merkel, E.; Flieder, D.; Pathak, H.; Koumbi, D.; Testa, J.R.; Eisenberg, B.; von Mehren, M.; Godwin, A.K. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc. Natl. Acad. Sci. USA 2008, 105, 8387–8392. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Johnstone, S.E.; Hemming, M.L.; Tarjan, D.R.; Hegazi, E.; Shareef, S.J.; Javed, N.M.; Raut, C.P.; Eschle, B.K.; et al. Altered chromosomal topology drives oncogenic programs in sdh-deficient gists. Nature 2019, 575, 229–233. [Google Scholar] [CrossRef]
- Mason, E.F.; Hornick, J.L. Conventional risk stratification fails to predict progression of succinate dehydrogenase-deficient gastrointestinal stromal tumors: A clinicopathologic study of 76 cases. Am. J. Surg. Pathol. 2016, 40, 1616–1621. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Carney, J.A. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (carney-stratakis syndrome): Molecular genetics and clinical implications. J. Intern. Med. 2009, 266, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Matyakhina, L.; Bei, T.A.; McWhinney, S.R.; Pasini, B.; Cameron, S.; Gunawan, B.; Stergiopoulos, S.G.; Boikos, S.; Muchow, M.; Dutra, A.; et al. Genetics of carney triad: Recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. J. Clin. Endocrinol. Metab. 2007, 92, 2938–2943. [Google Scholar] [CrossRef] [PubMed]
- Haller, F.; Moskalev, E.A.; Faucz, F.R.; Barthelmeß, S.; Wiemann, S.; Bieg, M.; Assie, G.; Bertherat, J.; Schaefer, I.M.; Otto, C.; et al. Aberrant DNA hypermethylation of sdhc: A novel mechanism of tumor development in carney triad. Endocr. Relat. Cancer 2014, 21, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.A. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (carney triad): Natural history, adrenocortical component, and possible familial occurrence. Mayo Clin. Proc. 1999, 74, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Killian, J.K.; Wang, Z.F.; Lasota, J.; Lau, C.; Jones, L.; Walker, R.; Pineda, M.; Zhu, Y.J.; Kim, S.Y.; et al. Immunohistochemical loss of succinate dehydrogenase subunit a (sdha) in gastrointestinal stromal tumors (gists) signals sdha germline mutation. Am. J. Surg. Pathol. 2013, 37, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Settas, N.; Faucz, F.R.; Stratakis, C.A. Succinate dehydrogenase (sdh) deficiency, carney triad and the epigenome. Mol. Cell. Endocrinol. 2018, 469, 107–111. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Antonescu, C.R.; DeMatteo, R.P.; Ganjoo, K.N.; Maki, R.G.; Pisters, P.W.; Raut, C.P.; Riedel, R.F.; Schuetze, S.; et al. Nccn task force report: Update on the management of patients with gastrointestinal stromal tumors. J. Natl. Compr. Cancer Netw. JNCCN 2010, 8 (Suppl. 2), S1–S41. [Google Scholar] [CrossRef]
- Murray, M.; Hatcher, H.; Jessop, F.; Williams, D.; Carroll, N.; Bulusu, R.; Judson, I. Treatment of wild-type gastrointestinal stromal tumor (wt-gist) with imatinib and sunitinib. Pediatric Blood Cancer 2008, 50, 386–388. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Morphology | KIT/DOG1 IHC | Diagnosis | Additional Positive IHC | Helpful Genetic Alteration(s) |
|---|---|---|---|---|
| Spindle cell | KIT+, DOG1+ | GIST | CD34 (70%), SMA (30%) | KIT, PDGFRA and others |
| KIT− (or very weak), DOG1- | Leiomyoma /Leiomyosarcoma | Desmin, SMA, caldesmon | ||
| Schwanomma | S100, SOX10 (nuclear) | |||
| Solitary fibrous tumor | CD34, STAT6 (nuclear) | NAB2-STAT6 | ||
| Fibromatosis | beta-catenin (nuclear) | CTNNB1 or APC mutation | ||
| IMT | ALK | ALK and ROS1 (rare) rearrangements | ||
| DDLPS | MDM2, CDK4 | MDM2, CDK4 amplification by FISH | ||
| Inflammatory fibroid polyp | CD34 | PDGFRA mutations | ||
| Epithelioid/mixed | KIT+, DOG1+ | GIST | SDHB retained/deficient (stomach) | KIT, PDGFRA, SDHA-D, SDHC promotor hypermethylation |
| KIT+/−, DOG1+/− | GIST | PDGFRA, KIT mutations | ||
| KIT−, DOG1− | PEComa | SMA, HMB45, MelanA, Desmin, TFE3 (subset of cases) | TSC2 mutation, TFE3-fusions | |
| Melanoma metastasis (can be KIT+) | SOX10, S100, HMB45, Melan A | cave: the common BRAFV600E mutation can be also found in GIST | ||
| Glomus tumor | SMA, caldesmon | NOTCH rearrangements and/or BRAF mutations (p.Val600Glu) | ||
| Neuroendocrine neoplasms | cytokeratin, synaptophysin, chromogranin A | DAXX, ATRX, p53, RB1 mutations |
| Genetic Type | Frequency | Anatomic Location | Treatment | |
|---|---|---|---|---|
| KIT mutations | ||||
| Exon 8 | <0.1% | |||
| Exon 9 | 6% | small & large bowel | Imatinib sensitiv (800 mg/d) | |
| Exon 11 | 66% | all locations | Imatinib sensitive | |
| Exon 13 | 1% | all locations | usually secondary mutation resistant to imatinib, responds to sunitinib | |
| Exon 17 | <1% | all locations | secondary mutation resistant to imatinib and sunitinib; have been shown to respond to other TKI like regorafenib | |
| PDGFRAmutations | ||||
| Exon 12 | 1% | all locations | ||
| Exon 14 | <1% | stomach | Imatinib sensitiv | |
| Exon 18 D842V | 6% | stomach | Imatinib/sunitinib resistant; good respons to avapritinib | |
| Exon 18 others | 1% | all locations | response to imatinib reported | |
| KIT/PDGFRA “wild-type” | Limited responses to imatinib Possible response to other TKIs (limited data) | |||
| SDHB IHC+/SDH-competent | NF1 mutation (assoc. with RD) | <1% | small bowel | |
| NRAS/HRAS/KRAS mutations | <1% | all locations (limited data) | ||
| BRAF mutation | 1% | most commonly stomach | ||
| Other rare mutations/fusions | all locations (limited data) | |||
| SDHB IHC−/SDH-deficient | SDHA/B/C/D mutations (CSS) | 2% | stomach | |
| Part of the CT * | 1% | stomach | ||
| SDHA mutation (young adults) | stomach | |||
| Sporadic pediatric wt- GIST | 1% | stomach |
| KIT Mutations | |
|---|---|
| Exon 8 | p.D419del |
| Exon 9 | p.K509I |
| Exon 11 | p.W557R p.V559A p.D579del p.V560G p.V560A p.L576P |
| Exon 13 | p.K642E p.N655K |
| Exon 17 | p.D820Y p.D820G p.N822Y |
| PDGFRA mutations | |
| Exon 12 | p.V561D p.Y555C |
| Exon 14 | p.P653L |
| Exon 18 | p.D842Y p.D846V |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brčić, I.; Argyropoulos, A.; Liegl-Atzwanger, B. Update on Molecular Genetics of Gastrointestinal Stromal Tumors. Diagnostics 2021, 11, 194. https://doi.org/10.3390/diagnostics11020194
Brčić I, Argyropoulos A, Liegl-Atzwanger B. Update on Molecular Genetics of Gastrointestinal Stromal Tumors. Diagnostics. 2021; 11(2):194. https://doi.org/10.3390/diagnostics11020194
Chicago/Turabian StyleBrčić, Iva, Alexandra Argyropoulos, and Bernadette Liegl-Atzwanger. 2021. "Update on Molecular Genetics of Gastrointestinal Stromal Tumors" Diagnostics 11, no. 2: 194. https://doi.org/10.3390/diagnostics11020194
APA StyleBrčić, I., Argyropoulos, A., & Liegl-Atzwanger, B. (2021). Update on Molecular Genetics of Gastrointestinal Stromal Tumors. Diagnostics, 11(2), 194. https://doi.org/10.3390/diagnostics11020194