
U1RNP/lncRNA/Transcription Cycle Axis Promotes Tumorigenesis of Hepatocellular Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Identification of U1-lncRNAs and Correlated mRNAs
2.3. Cellular Location of lncRNAs with U1-Binding Motif
2.4. Functional Annotation and Regulation Network Construction
2.5. Construction of the Prognostic Based on U1-lncRNAs
2.6. Cell Culture and Transfection
2.7. Cell Proliferative Assay
2.8. Cell Cycle Analysis
2.9. Quantitative Real-Time Polymerase Chain Reaction
2.10. Western Blot
2.11. Statistical Analysis
3. Results
3.1. Overview of U1RNPs and Its Related lncRNAs in HCC
3.2. Distribution of HCC-Specific lncRNAs with U1 Binding Sites
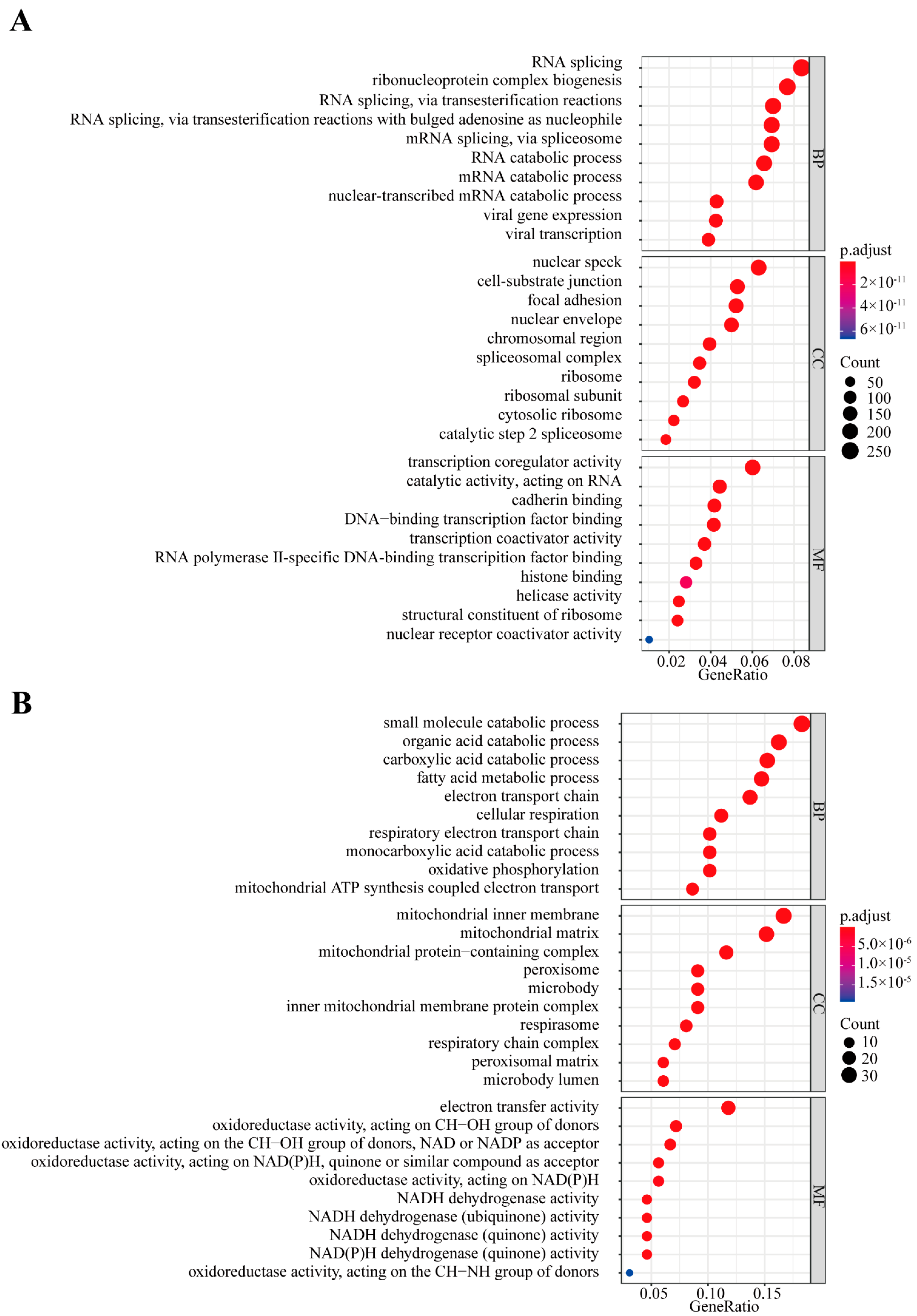
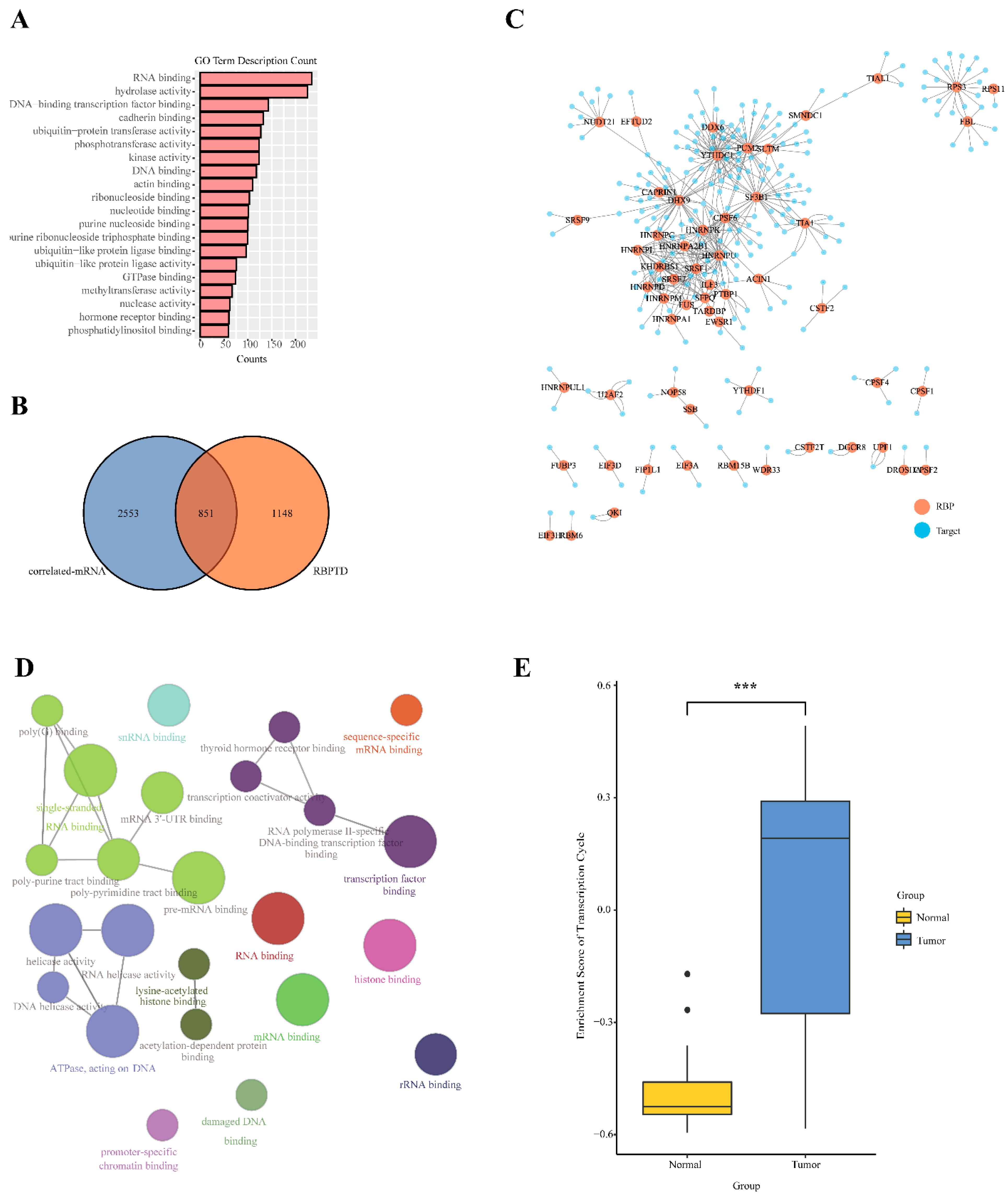
3.3. Function and Network Analysis of U1-lncRNAs and Correlated mRNAs
3.4. U1-lncRNAs Activates the Transcription Cycle to Promote HCC
3.5. Construction and Validation of the U1-lncRNAs-Based Prognostic Model
3.6. Biological Function of SNRPD2 in HCC Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, A. Hepatocellular Carcinoma. New Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [CrossRef] [Green Version]
- Wilkinson, M.; Charenton, C.; Nagai, K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. [Google Scholar] [CrossRef]
- Kaida, D.; Berg, M.G.; Younis, I.; Kasim, M.; Singh, L.N.; Wan, L.; Dreyfuss, G. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 2010, 468, 664–668. [Google Scholar] [CrossRef]
- Oh, J.-M.; Venters, C.C.; Di, C.; Pinto, A.M.; Wan, L.; Younis, I.; Cai, Z.; Arai, C.; So, B.R.; Duan, J.; et al. U1 snRNP regulates cancer cell migration and invasion in vitro. Nat. Commun. 2020, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Lu, J.Y.; Zhang, X.; Shao, W.; Xu, Y.; Li, P.; Hong, Y.; Cui, L.; Shan, G.; Tian, B.; et al. U1 snRNP regulates chromatin retention of noncoding RNAs. Nature 2020, 580, 147–150. [Google Scholar] [CrossRef]
- Wong, C.M.; Tsang, F.H.; Ng, I.O. Non-coding RNAs in hepatocellular carcinoma: Molecular functions and pathological implications. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 137–151. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.M.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- Grinchuk, O.V.; Yenamandra, S.P.; Iyer, R.; Singh, M.; Lee, H.K.; Lim, K.H.; Chow, P.K.; Kuznetsov, V.A. Tumor-adjacent tissue co-expression profile analysis reveals pro-oncogenic ribosomal gene signature for prognosis of resectable hepatocellular carcinoma. Mol. Oncol. 2018, 12, 89–113. [Google Scholar] [CrossRef] [Green Version]
- Sheth, N.; Roca, X.; Hastings, M.; Roeder, T.; Krainer, A.; Sachidanandam, R. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res. 2006, 34, 3955–3967. [Google Scholar] [CrossRef] [Green Version]
- Busch, A.; Hertel, K.J. HEXEvent: A database of human EXon splicing events. Nucleic Acids Res. 2013, 41, D118–D124. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [Green Version]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Hermoso Pulido, T.; Guigo, R.; Johnson, R. LncATLAS database for subcellular localization of long noncoding RNAs. RNA 2017, 23, 1080–1087. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Guo, Z.W.; Zhai, X.M.; Yang, X.X.; Wu, Y.S.; Liu, T.C. RBPTD: A database of cancer-related RNA-binding proteins in humans. Database 2020, 2020, baz156. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Teng, X.; Chen, X.; Xue, H.; Tang, Y.; Zhang, P.; Kang, Q.; Hao, Y.; Chen, R.; Zhao, Y.; He, S. NPInter v4.0: An integrated database of ncRNA interactions. Nucleic Acids Res. 2020, 48, D160–D165. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, S.; Zhu, Y.; Xi, X.; Bao, P.; Ma, Z.; Kapral, T.H.; Chen, S.; Zagrovic, B.; Yang, Y.T.; et al. POSTAR3: An updated platform for exploring post-transcriptional regulation coordinated by RNA-binding proteins. Nucleic Acids Res. 2022, 50, D287–D294. [Google Scholar] [CrossRef] [PubMed]
- Vervoort, S.J.; Devlin, J.R.; Kwiatkowski, N.; Teng, M.; Gray, N.S.; Johnstone, R.W. Targeting transcription cycles in cancer. Nat. Rev. Cancer 2022, 22, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [Green Version]
- Shuai, S.; Suzuki, H.; Diaz-Navarro, A.; Nadeu, F.; Kumar, S.; Gutierrez-Fernandez, A.; Delgado, J.; Pinyol, M.; López-Otín, C.; Puente, X.S.; et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 2019, 574, 712–716. [Google Scholar] [CrossRef]
- Suzuki, H.; Kumar, S.; Shuai, S.; Diaz-Navarro, A.; Gutierrez-Fernandez, A.; de Antonellis, P.; Cavalli, F.M.G.; Juraschka, K.; Farooq, H.; Shibahara, I.; et al. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 2019, 574, 707–711. [Google Scholar] [CrossRef]
- Jia, D.; Wei, L.; Guo, W.; Zha, R.; Bao, M.; Chen, Z.; Zhao, Y.; Ge, C.; Zhao, F.; Chen, T.; et al. Genome-wide copy number analyses identified novel cancer genes in hepatocellular carcinoma. Hepatology 2011, 54, 1227–1236. [Google Scholar] [CrossRef]
- Zhan, Y.T.; Li, L.; Zeng, T.T.; Zhou, N.N.; Guan, X.Y.; Li, Y. SNRPB-mediated RNA splicing drives tumor cell proliferation and stemness in hepatocellular carcinoma. Aging 2020, 13, 537–554. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, J.; Zuo, D.; Yuan, Y.; Qiu, Y.; Qiao, L.; He, W.; Li, B.; Yuan, Y. SNRPC promotes hepatocellular carcinoma cell motility by inducing epithelial-mesenchymal transition. FEBS Open Bio. 2021, 11, 1757–1770. [Google Scholar] [CrossRef]
- Cai, J.; Zhou, M.; Xu, J. N6-methyladenosine (m6A) RNA methylation regulator SNRPC is a prognostic biomarker and is correlated with immunotherapy in hepatocellular carcinoma. World J. Surg. Oncol. 2021, 19, 241. [Google Scholar] [CrossRef]
- Tamura, K.; Furihata, M.; Tsunoda, T.; Ashida, S.; Takata, R.; Obara, W.; Yoshioka, H.; Daigo, Y.; Nasu, Y.; Kumon, H.; et al. Molecular features of hormone-refractory prostate cancer cells by genome-wide gene expression profiles. Cancer Res. 2007, 67, 5117–5125. [Google Scholar] [CrossRef] [Green Version]
- Genovesi, L.A.; Millar, A.; Tolson, E.; Singleton, M.; Hassall, E.; Kojic, M.; Brighi, C.; Girard, E.; Andradas, C.; Kuchibhotla, M.; et al. Systems pharmacogenomics identifies novel targets and clinically actionable therapeutics for medulloblastoma. Genome Med. 2021, 13, 103. [Google Scholar] [CrossRef]
- Zhu, P.; Wang, Y.; Wu, J.; Huang, G.; Liu, B.; Ye, B.; Du, Y.; Gao, G.; Tian, Y.; He, L.; et al. LncBRM initiates YAP1 signalling activation to drive self-renewal of liver cancer stem cells. Nat. Commun. 2016, 7, 13608. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Huo, X.; Yang, X.-R.; He, J.; Cheng, L.; Wang, N.; Deng, X.; Jingyuan, F.; Wang, N.; Wang, C.; et al. STAT3-mediated upregulation of lncRNA HOXD-AS1 as a ceRNA facilitates liver cancer metastasis by regulating SOX4. Mol. Cancer 2017, 16, 136. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, L.; Chen, T.; Liu, X.; Guo, Y.; Zhu, Q.; Tong, X.; Yang, W.; Xu, Q.; Huang, D.; et al. A novel lncRNA MCM3AP-AS1 promotes the growth of hepatocellular carcinoma by targeting miR-194-5p/FOXA1 axis. Mol. Cancer 2019, 18, 28. [Google Scholar] [CrossRef]
- Hu, J.-C.; Wang, S.-S.; Chou, Y.-E.; Chiu, K.-Y.; Li, J.-R.; Chen, C.-S.; Hung, S.-C.; Yang, C.-K.; Ou, Y.-C.; Cheng, C.-L.; et al. Associations between LncRNA MALAT1 polymorphisms and lymph node metastasis in prostate cancer. Diagnostics 2021, 11, 1692. [Google Scholar] [CrossRef]
- Lin, K.; Song, L.J.; Ma, J.; Zhang, T.S.; You, D.Y.; He, Y.W. Identification of cancer hallmark-associated gene and lncRNA cooperative regulation pairs and dictate lncRNA roles in oral squamous cell carcinoma. J. Cell. Mol. Med. 2020, 24, 5213–5223. [Google Scholar] [CrossRef] [Green Version]
- Desterro, J.; Bak-Gordon, P.; Carmo-Fonseca, M. Targeting mRNA processing as an anticancer strategy. Nat. Rev. Drug Discov. 2020, 19, 112–129. [Google Scholar] [CrossRef]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef]
- Xu, Y.; Vakoc, C.R. Targeting Cancer Cells with BET Bromodomain Inhibitors. Cold Spring Harb. Perspect. Med. 2017, 7, a026674. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zuroske, T.; Watts, J.K. RNA therapeutics on the rise. Nat. Rev. Drug Discov. 2020, 19, 441–442. [Google Scholar] [CrossRef]
- Liu, S.J.; Dang, H.X.; Lim, D.A.; Feng, F.Y.; Maher, C.A. Long noncoding RNAs in cancer metastasis. Nat. Rev. Cancer 2021, 21, 446–460. [Google Scholar] [CrossRef]
- Winkle, M.; El-Daly, S.; Fabbri, M.; Calin, G. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef]
- Lin, C.; Yang, L. Long noncoding RNA in cancer: Wiring signaling circuitry. Trends Cell Biol. 2018, 28, 287–301. [Google Scholar] [CrossRef]
- Jourdain, A.A.; Begg, B.E.; Mick, E.; Shah, H.; Calvo, S.E.; Skinner, O.S.; Sharma, R.; Blue, S.M.; Yeo, G.W.; Burge, C.B.; et al. Loss of LUC7L2 and U1 snRNP subunits shifts energy metabolism from glycolysis to OXPHOS. Mol. Cell 2021, 81, 1905–1919. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Zhang, S.; Huang, M.; Hu, H.; Xie, Y. U1RNP/lncRNA/Transcription Cycle Axis Promotes Tumorigenesis of Hepatocellular Carcinoma. Diagnostics 2022, 12, 1133. https://doi.org/10.3390/diagnostics12051133
Li S, Zhang S, Huang M, Hu H, Xie Y. U1RNP/lncRNA/Transcription Cycle Axis Promotes Tumorigenesis of Hepatocellular Carcinoma. Diagnostics. 2022; 12(5):1133. https://doi.org/10.3390/diagnostics12051133
Chicago/Turabian StyleLi, Shun, Shuaiyin Zhang, Mingle Huang, Huanjing Hu, and Yubin Xie. 2022. "U1RNP/lncRNA/Transcription Cycle Axis Promotes Tumorigenesis of Hepatocellular Carcinoma" Diagnostics 12, no. 5: 1133. https://doi.org/10.3390/diagnostics12051133