Single-Cell RNA Sequencing Analysis Reveals Metabolic Changes in Epithelial Glycosphingolipids and Establishes a Prognostic Risk Model for Pancreatic Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Single-Cell Sequencing Data Quality Control and Processing
2.2. Single-Cell Metabolic Analysis
2.3. Risk Model Construction
2.4. Risk Model Evaluation and Validation
2.5. Nomogram Model Construction
2.6. Mutation Landscape Analysis
2.7. Immune Cell Infiltration Estimation and Immune Subtype Analysis
2.8. Drug Response Prediction
2.9. Statistical Analysis
3. Results
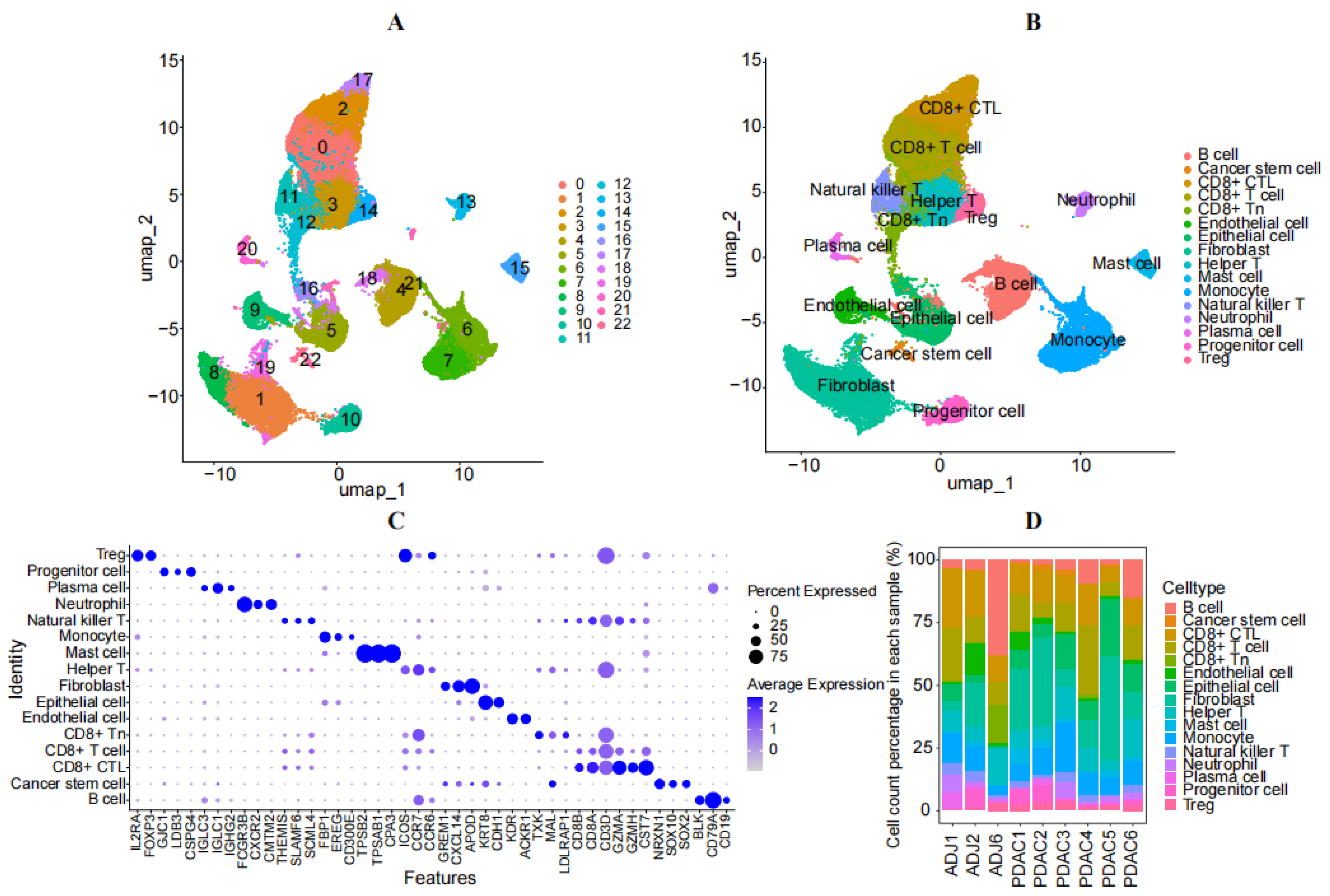
3.1. Analysis of Single-Cell Sequencing
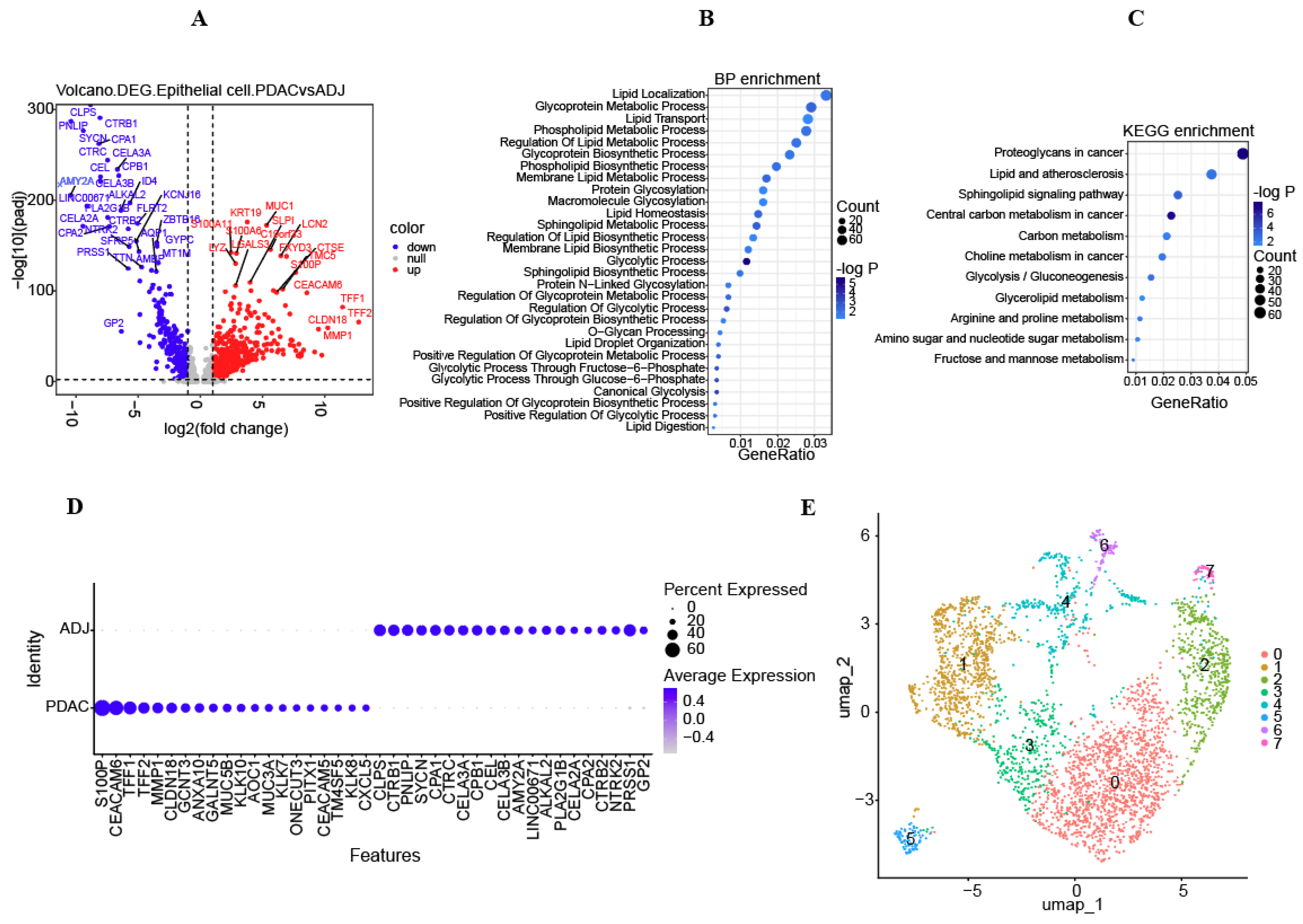
3.2. Metabolic Characteristics of Epithelial Cells and Screened DEGs
3.3. Development, Assessment, and Verification of Risk Models
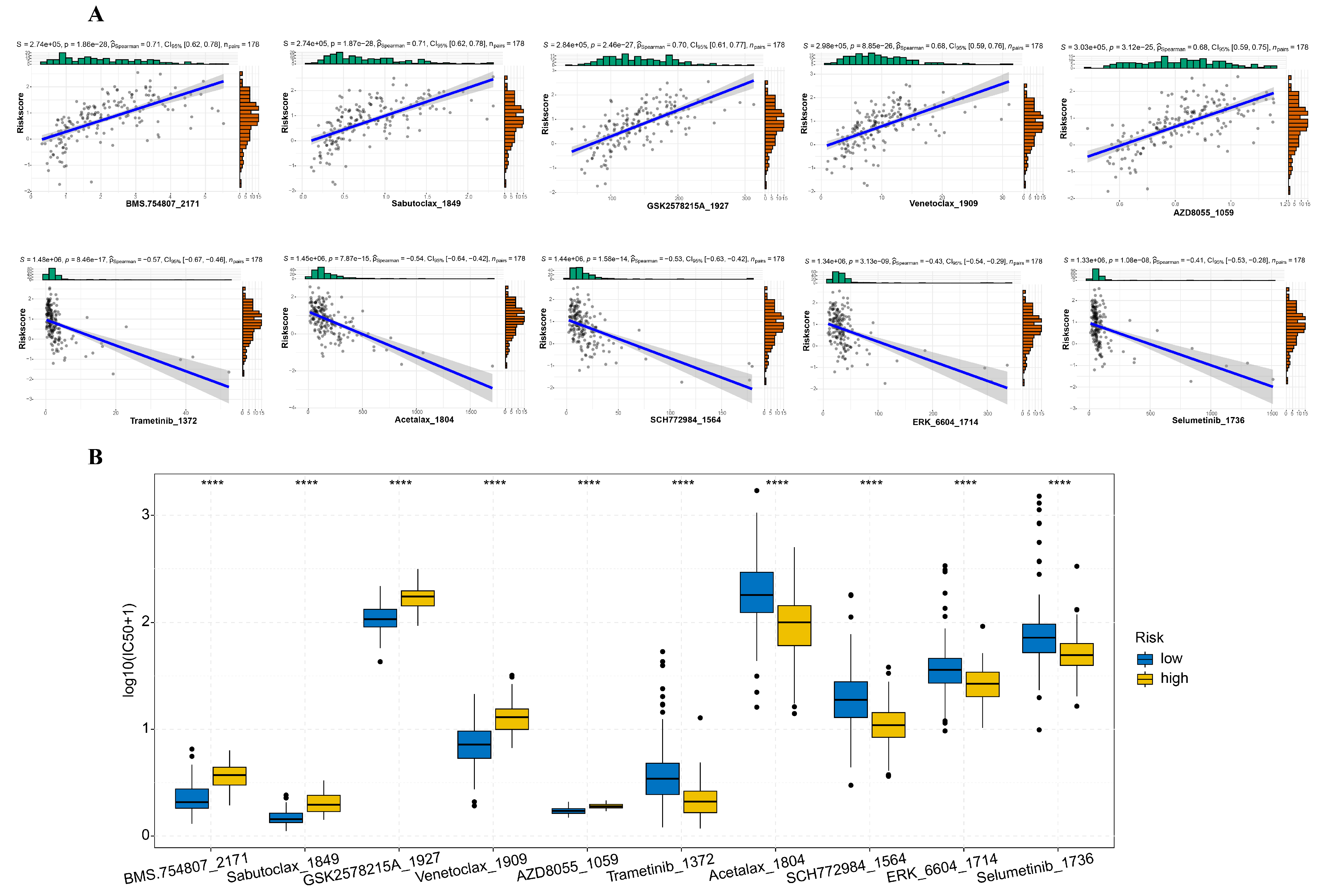
3.4. Mutation Profile and Drug Sensitivity Variances between High-Risk and Low-Risk Cohorts
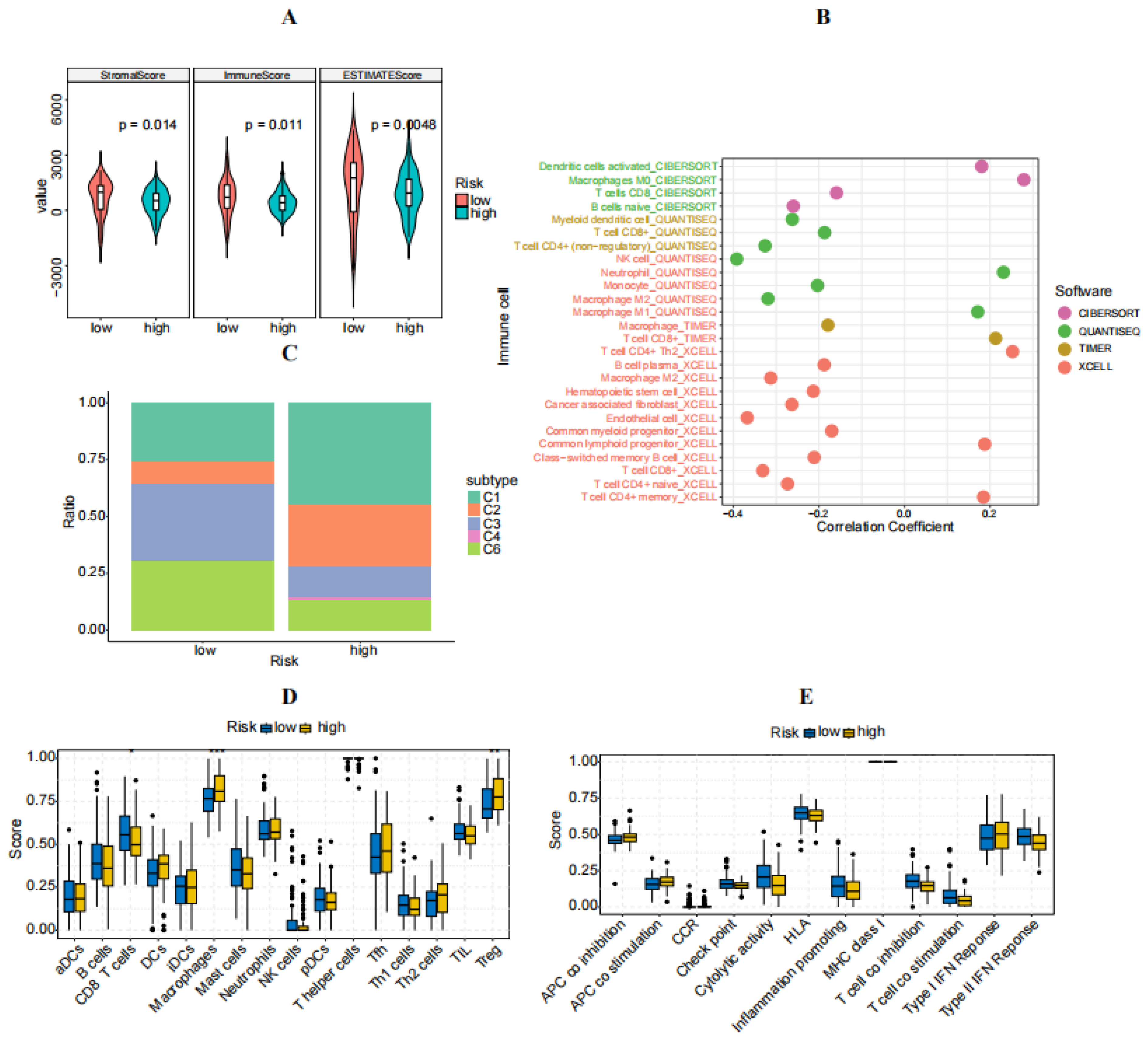
3.5. Immune Characteristics of High- and Low-Risk Groups
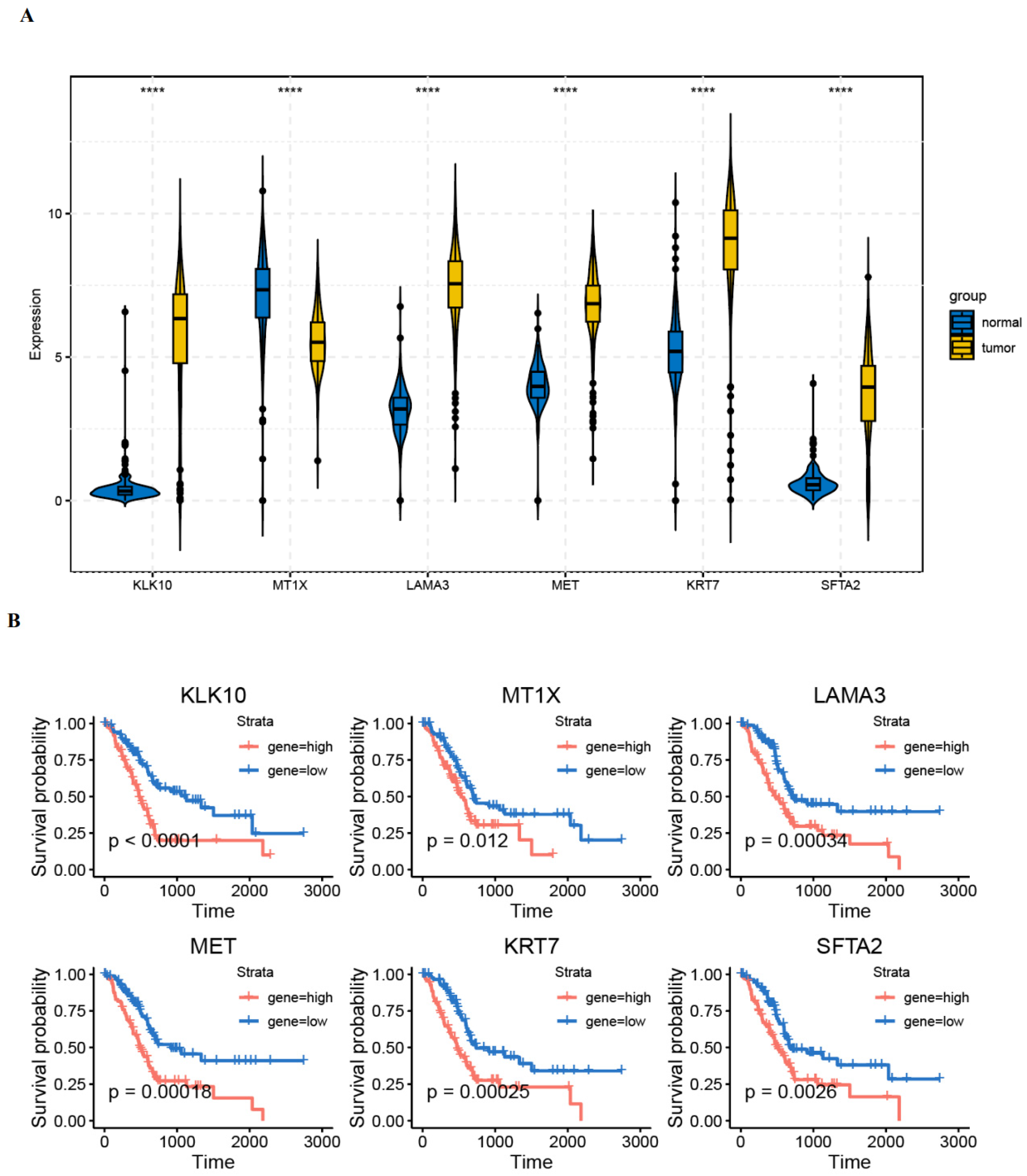
3.6. Validation of Gene Expression in PAAD Tissues
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Gallick, G.E. Gemcitabine resistance in pancreatic cancer: Picking the key players. Clin. Cancer Res. 2008, 14, 1284–1285. [Google Scholar] [CrossRef] [PubMed]
- Gyamfi, J.; Kim, J.; Choi, J. Cancer as a Metabolic Disorder. Int. J. Mol. Sci. 2022, 23, 1155. [Google Scholar] [CrossRef] [PubMed]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S.; et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit. Rev. Oncol. Hematol. 2017, 114, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Gunda, V.; Souchek, J.; Abrego, J.; Shukla, S.K.; Goode, G.D.; Vernucci, E.; Dasgupta, A.; Chaika, N.V.; King, R.J.; Li, S.; et al. MUC1-Mediated Metabolic Alterations Regulate Response to Radiotherapy in Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 5881–5891. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Karasinska, J.M.; Topham, J.T.; Kalloger, S.E.; Jang, G.H.; Denroche, R.E.; Culibrk, L.; Williamson, L.M.; Wong, H.L.; Lee, M.K.C.; O’Kane, G.M.; et al. Altered Gene Expression along the Glycolysis-Cholesterol Synthesis Axis Is Associated with Outcome in Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Mehla, K.; Singh, P.K. Metabolic Subtyping for Novel Personalized Therapies Against Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Grippo, P.J.; Munshi, H.G. (Eds.) Pancreatic Cancer and Tumor Microenvironment; Transworld Research Network: Trivandrum, India, 2012. [Google Scholar]
- Cumin, C.; Huang, Y.L.; Everest-Dass, A.; Jacob, F. Deciphering the Importance of Glycosphingolipids on Cellular and Molecular Mechanisms Associated with Epithelial-to-Mesenchymal Transition in Cancer. Biomolecules 2021, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Quek, L.E.; Sultani, G.; Turner, N. Epithelial-mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Cancer Metab. 2016, 4, 19. [Google Scholar] [CrossRef]
- Yang, J.; Ren, B.; Yang, G.; Wang, H.; Chen, G.; You, L.; Zhang, T.; Zhao, Y. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell. Mol. Life Sci. 2020, 77, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Bernard, V.; Semaan, A.; Monberg, M.E.; Huang, J.; Stephens, B.M.; Lin, D.; Rajapakshe, K.I.; Weston, B.R.; Bhutani, M.S.; et al. Elucidation of Tumor-Stromal Heterogeneity and the Ligand-Receptor Interactome by Single-Cell Transcriptomics in Real-world Pancreatic Cancer Biopsies. Clin. Cancer Res. 2021, 27, 5912–5921. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, S.; Ma, J.; Chen, Z.; Song, G.; Rao, D.; Cheng, Y.; Huang, S.; Liu, Y.; Jiang, S.; et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level. Cancer Discov. 2022, 12, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, G.; Capasso, S.; Sticco, L.; Russo, D. Glycosphingolipids: Synthesis and functions. FEBS J. 2013, 280, 6338–6353. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.L.; Hung, J.T.; Ho, M.Y.; Yu, J. Alterations of Glycosphingolipids in Embryonic Stem Cell Differentiation and Development of Glycan-Targeting Cancer Immunotherapy. Stem Cells Dev. 2016, 25, 1532–1548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ji, S.; Zhang, X.; Lu, M.; Hu, Y.; Han, Y.; Shui, G.; Lam, S.M.; Zou, X. Human CPTP promotes growth and metastasis via sphingolipid metabolite ceramide and PI4KA/AKT signaling in pancreatic cancer cells. Int. J. Biol. Sci. 2022, 18, 4963–4983. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Guan, F.; Handa, K.; Hakomori, S.I. Specific glycosphingolipids mediate epithelial-to-mesenchymal transition of human and mouse epithelial cell lines. Proc. Natl. Acad. Sci. USA 2009, 106, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Chung, T.W.; Choi, H.J.; Kwak, C.H.; Song, K.H.; Suh, S.J.; Kwon, K.M.; Chang, Y.C.; Park, Y.G.; Chang, H.W.; et al. Ganglioside GM3 participates in the TGF-beta1-induced epithelial-mesenchymal transition of human lens epithelial cells. Biochem. J. 2013, 449, 241–251. [Google Scholar] [CrossRef]
- Cao, X.Y.; Zhang, X.X.; Yang, M.W.; Hu, L.P.; Jiang, S.H.; Tian, G.A.; Zhu, L.L.; Li, Q.; Sun, Y.W.; Zhang, Z.G. Aberrant upregulation of KLK10 promotes metastasis via enhancement of EMT and FAK/SRC/ERK axis in PDAC. Biochem. Biophys. Res. Commun. 2018, 499, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wu, X.; Wang, S.; Liu, S.; Gao, X. Spatial heterogeneity and Immune infiltration of cellular lysosomal pathways reveals a new blueprint for tumor heterogeneity in esophageal cancer. Front. Endocrinol. 2023, 14, 1138457. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.; Balasubramanian, B.; Venkatraman, S.; Thummarati, P.; Tunganuntarat, J.; Phueakphud, N.; Kanjanasirirat, P.; Khumpanied, T.; Kongpracha, P.; Kittirat, Y.; et al. Upregulated LAMA3 modulates proliferation, adhesion, migration and epithelial-to-mesenchymal transition of cholangiocarcinoma cells. Sci. Rep. 2023, 13, 22598. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M.; Trusolino, L.; Boccaccio, C. Known and novel roles of the MET oncogene in cancer: A coherent approach to targeted therapy. Nat. Rev. Cancer 2018, 18, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Bauer, C.A.; Ohlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut 2019, 68, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Harbaum, L.; Pollheimer, M.J.; Kornprat, P.; Lindtner, R.A.; Schlemmer, A.; Rehak, P.; Langner, C. Keratin 7 expression in colorectal cancer—Freak of nature or significant finding? Histopathology 2011, 59, 225–234. [Google Scholar] [CrossRef]
- Lambaudie, E.; Chereau, E.; Pouget, N.; Thomassin, J.; Minsat, M.; Charafe-Jauffret, E.; Jacquemier, J.; Houvenaeghel, G. Cytokeratin 7 as a predictive factor for response to concommitant radiochemotherapy for locally advanced cervical cancer: A preliminary study. Anticancer Res. 2014, 34, 177–181. [Google Scholar]
- Atay, S. Integrated transcriptome meta-analysis of pancreatic ductal adenocarcinoma and matched adjacent pancreatic tissues. PeerJ 2020, 8, e10141. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, X.; Zhang, Q.; Zhang, T.; Xie, J.; Wei, W.; Wang, Y.; Yu, H.; Zhou, H. A necroptosis related prognostic model of pancreatic cancer based on single cell sequencing analysis and transcriptome analysis. Front. Immunol. 2022, 13, 1022420. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Q.; Liu, X.; Wang, F.; Ma, Y.; Zhang, S.; Shao, Z.; Yang, Y.; Tian, X. Single Cell RNA-Seq Identifies Immune-Related Prognostic Model and Key Signature-SPP1 in Pancreatic Ductal Adenocarcinoma. Genes 2022, 13, 1760. [Google Scholar] [CrossRef]
- Liu, Q.; Li, R.; Wu, H.; Liang, Z. A novel cuproptosis-related gene model predicts outcomes and treatment responses in pancreatic adenocarcinoma. BMC Cancer 2023, 23, 226. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ba, Q.; Wang, X.; Hu, H.; Lu, Y. Single-Cell RNA Sequencing Analysis Reveals Metabolic Changes in Epithelial Glycosphingolipids and Establishes a Prognostic Risk Model for Pancreatic Cancer. Diagnostics 2024, 14, 1094. https://doi.org/10.3390/diagnostics14111094
Ba Q, Wang X, Hu H, Lu Y. Single-Cell RNA Sequencing Analysis Reveals Metabolic Changes in Epithelial Glycosphingolipids and Establishes a Prognostic Risk Model for Pancreatic Cancer. Diagnostics. 2024; 14(11):1094. https://doi.org/10.3390/diagnostics14111094
Chicago/Turabian StyleBa, Qinwen, Xiong Wang, Hui Hu, and Yanjun Lu. 2024. "Single-Cell RNA Sequencing Analysis Reveals Metabolic Changes in Epithelial Glycosphingolipids and Establishes a Prognostic Risk Model for Pancreatic Cancer" Diagnostics 14, no. 11: 1094. https://doi.org/10.3390/diagnostics14111094