
Genetic and Phenotypic Spectrum of KMT2D Variants in Taiwanese Case Series of Kabuki Syndrome
 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Group and Data Collection
2.2. Molecular Analysis
2.3. Study Limitations
2.4. Variant Classification
3. Results
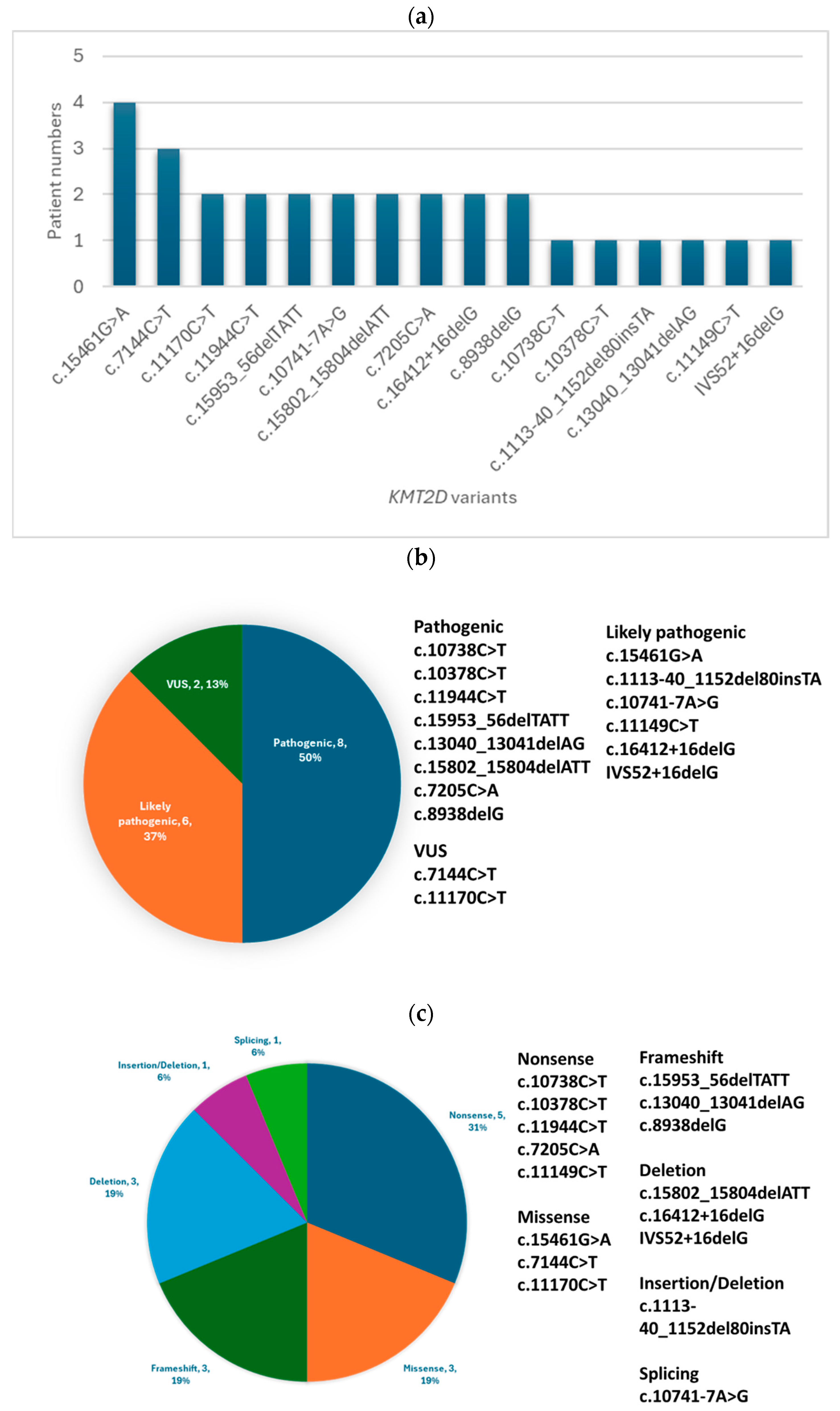
3.1. Molecular Findings
3.2. Clinical Features
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adam, M.P.; Banka, S.; Bjornsson, H.T.; Bodurtha, J.; Chudley, A.E.; Harris, J.; Kawame, H.; Lanpher, B.C.; Lindsley, A.W.; Merla, G.; et al. Kabuki syndrome: International consensus diagnostic criteria. J. Med. Genet. 2019, 56, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Usluer, E.; Sayın, G.Y.; Güneş, N.; Kasap, B.; Tüysüz, B. Investigation of genetic and phenotypic heterogeneity in 37 Turkish patients with Kabuki and Kabuki-like phenotype. Am. J. Med. Genet. A 2022, 188, 2976–2987. [Google Scholar] [CrossRef] [PubMed]
- Niikawa, N.; Kuroki, Y.; Kajii, T.; Matsuura, N.; Ishikiriyama, S.; Tonoki, H.; Ishikawa, N.; Yamada, Y.; Fujita, M.; Umemoto, H.; et al. Kabuki make-up (Niikawa-Kuroki) syndrome: A study of 62 patients. Am. J. Med. Genet. 1988, 31, 565–589. [Google Scholar] [CrossRef] [PubMed]
- Niikawa, N.; Matsuura, N.; Fukushima, Y.; Ohsawa, T.; Kajii, T. Kabuki make-up syndrome: A syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J. Pediatr. 1981, 99, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.; Grisart, B.; Digilio, M.C.; Benoit, V.; Crespin, M.; Ghariani, S.C.; Maystadt, I.; Dallapiccola, B.; Verellen-Dumoulin, C. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am. J. Hum. Genet. 2012, 90, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Bögershausen, N.; Gatinois, V.; Riehmer, V.; Kayserili, H.; Becker, J.; Thoenes, M.; Simsek-Kiper, P.Ö.; Barat-Houari, M.; Elcioglu, N.H.; Wieczorek, D.; et al. Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2. Hum. Mutat. 2016, 37, 847–864. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Koshimizu, E.; Okamoto, N.; Mizuno, S.; Ogata, T.; Nagai, T.; Kosho, T.; Ohashi, H.; Kato, M.; Sasaki, G.; et al. MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am. J. Med. Genet. A 2013, 161A, 2234–2243. [Google Scholar] [CrossRef] [PubMed]
- Banka, S.; Veeramachaneni, R.; Reardon, W.; Howard, E.; Bunstone, S.; Ragge, N.; Parker, M.J.; Crow, Y.J.; Kerr, B.; Kingston, H.; et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur. J. Hum. Genet. 2012, 20, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Bögershausen, N.; Tsai, I.C.; Pohl, E.; Kiper, P.Ö.S.; Beleggia, F.; Percin, E.F.; Keupp, K.; Matchan, A.; Milz, E.; Alanay, Y.; et al. RAP1-mediated MEK/ERK pathway defects in Kabuki syndrome. J. Clin. Investig. 2015, 125, 3585–3599. [Google Scholar] [CrossRef] [PubMed]
- Lange, L.; Pagnamenta, A.T.; Lise, S.; Clasper, S.; Stewart, H.; Akha, E.S.; Quaghebeur, G.; Knight, S.J.L.; Keays, D.A.; Taylor, J.C.; et al. A de novo frameshift in HNRNPK causing a Kabuki-like syndrome with nodular heterotopia. Clin. Genet. 2016, 90, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Au, P.Y.B.; You, J.; Caluseriu, O.; Schwartzentruber, J.; Majewski, J.; Bernier, F.P.; Ferguson, M.; Valle, D.; Parboosingh, J.S.; Sobreira, N.; et al. GeneMatcher aids in the identification of a new malformation syndrome with intellectual disability, unique facial dysmorphisms, and skeletal and connective tissue abnormalities caused by de novo variants in HNRNPK. Hum. Mutat. 2015, 36, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Makrythanasis, P.; van Bon, B.W.; Steehouwer, M.; Rodríguez-Santiago, B.; Simpson, M.; Dias, P.; Anderlid, B.M.; Arts, P.; Bhat, M.; Augello, B.; et al. MLL2 mutation detection in 86 patients with Kabuki syndrome: A genotype-phenotype study. Clin. Genet. 2013, 84, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Lehman, N.; Mazery, A.C.; Visier, A.; Baumann, C.; Lachesnais, D.; Capri, Y.; Toutain, J.; Dieux, A.; Sanlaville, D.; Odent, S.; et al. Molecular, clinical and neuropsychological study in 31 patients with Kabuki syndrome and KMT2D mutations. Clin. Genet. 2017, 92, 298–305. [Google Scholar] [CrossRef] [PubMed]
- So, P.L.; Luk, H.M.; Yu, K.P.T.; Ma, A.S.; Ting, C.T.; Liu, A.P.Y.; Hung, K.L.Y.; Chu, Y.W.; Lo, F.M.; Chow, P.C.W.; et al. Clinical and molecular characterization study of Chinese Kabuki syndrome in Hong Kong. Am. J. Med. Genet. A 2021, 185, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Priolo, M.; Micale, L.; Augello, B.; Fusco, C.; Zucchetti, F.; Prontera, P.; Paduano, V.; Biamino, E.; Selicorni, A.; Mammi, I.; et al. Absence of deletion and duplication of MLL2 and KDM6A genes in a large cohort of patients with Kabuki syndrome. Mol. Genet. Metab. 2012, 107, 627–629. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Patient | Sex | KMT2D cDNA Change | KMT2D Amino Acid Change | Pathogenicity | ACMG Classification | Inheritance |
|---|---|---|---|---|---|---|
| 1 | M | c.15461G>A | p.Arg5154Gln | Missense | Likely Pathogenic | De novo |
| 2 | M | c.10738C>T | p.Gln3580 * | Nonsense | Pathogenic | De novo |
| 3 | M | c.10378C>T | p.Gln3460 * | Nonsense | Pathogenic | De novo |
| 4 | M | c.11944C>T | p.Arg3982 * | Nonsense | Pathogenic | De novo |
| 5 | M | c.1113-40_1152del80insTA | - | Insertion/ Deletion | Likely Pathogenic | Unknown |
| 6 | F | c.15953_56delTATT | p.Leu5318Serfs *14 | Frameshift | Pathogenic | De novo |
| 7 | F | c.13040_13041delAG | p.Gln4347Argfs *24 | Frameshift | Pathogenic | De novo |
| c.7144C>T | p.Pro2382Ser | Missense | Uncertain Significance | Unknown | ||
| 8 | M | c.10741-7A>G | - | Splicing | Likely Pathogenic | Unknown |
| c.15802_15804delATT | p.Ile5268del | Deletion | Pathogenic | De novo | ||
| 9 | F | c.7205C>A | p.Ser2402 * | Nonsense | Pathogenic | De novo |
| 10 | F | c.15802_15804delATT | p.Ile5268del | Deletion | Pathogenic | De novo |
| 11 | M | c.7205C>A | p.Ser2402 * | Nonsense | Pathogenic | De novo |
| 12 | F | c.15953_56delTATT | p.Leu5318Serfs *14 | Frameshift | Pathogenic | De novo |
| 13 | M | c.15461G>A | p.Arg5154Gln | Missense | Likely Pathogenic | De novo |
| 14 | M | c.11149C>T | p.Gln3717 * | Nonsense | Likely Pathogenic | De novo |
| 15 | F | c.7144C>T | p.Pro2382Ser | Missense | Uncertain Significance | Unknown |
| 16 | F | c.7144C>T | p.Pro2382Ser | Missense | Uncertain Significance | Unknown |
| c.16412+16delG | - | Deletion | Likely Pathogenic | Unknown | ||
| 17 | F | c.15461G>A | p.Arg5154Gln | Missense | Likely Pathogenic | De novo |
| 18 | M | c.8938delG | p.Ala2980Profs *24 | Frameshift | Pathogenic | De novo |
| 19 | M | c.11170C>T | p.Pro2382Ser | Missense | Uncertain Significance | Unknown |
| c.16412+16delG | - | Deletion | Likely Pathogenic | Unknown | ||
| 20 | F | c.8938delG | p.Ala2980Profs *24 | Frameshift | Pathogenic | De novo |
| 21 | F | c.15461G>A | p.Arg5154Gln | Missense | Likely Pathogenic | De novo |
| 22 | F | c.16412+16delG | - | Deletion | Likely Pathogenic | Unknown |
| 23 | M | c.10741-7A>G | - | Splicing | Likely Pathogenic | Unknown |
| Clinical Feature | Number of Patients (Total = 23) | Percentage (%) |
|---|---|---|
| Distinct facial features | 23 | 100 |
| Intellectual disability | 23 | 100 |
| Developmental delay | 22 | 95.7 |
| Speech delay | 18 | 78.3 |
| Hypotonia | 16 | 69.6 |
| Congenital heart defects | 16 | 69.6 |
| ASD, ostium secundum | 6/16 | 37.5 |
| VSD, perimembranous subaortic | 3/16 | 18.8 |
| Aortic coarctation | 3/16 | 18.8 |
| Bicuspid aortic valve | 2/16 | 12.5 |
| Persistent left superior vena cava | 2/16 | 12.5 |
| Mitral valve prolapse | 2/16 | 12.5 |
| Mitral regurgitation | 2/16 | 12.5 |
| Patent ductus arteriosus | 2/16 | 12.5 |
| Mitral stenosis | 1/16 | 6.3 |
| Aberrant right subclavian artery | 1/16 | 6.3 |
| Vascular ring | 1/16 | 6.3 |
| Dilated aortic root | 1/16 | 6.3 |
| Interrupted aortic arch | 1/16 | 6.3 |
| Subaortic ridge | 1/16 | 6.3 |
| Left isomerism of the heart | 1/16 | 6.3 |
| Aortic dissection | 1/16 | 6.3 |
| Redundant mitral valve | 1/16 | 6.3 |
| Redundant tricuspid valve | 1/16 | 6.3 |
| Recurrent infections | 15 | 65.2 |
| Short stature | 14 | 60.9 |
| Feeding difficulties | 10 | 43.5 |
| Hearing impairment | 9 | 39.1 |
| Seizures | 6 | 26.1 |
| Cleft palate | 6 | 26.1 |
| Renal anomalies | 5 | 21.7 |
| Preauricular pits | 4 | 17.4 |
| Ophthalmologic abnormalities | 4 | 17.4 |
| Sacral dimple | 2 | 8.7 |
| Cryptorchidism | 1 | 4.3 |
| Hip dislocation | 1 | 4.3 |
| Premature thelarche | 1 | 4.3 |
| Gastrointestinal anomalies | 1 | 4.3 |
| Dental anomalies | 1 | 4.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-L.; Chuang, C.-K.; Chen, M.-R.; Lin, J.-L.; Chiu, H.-C.; Chang, Y.-H.; Tu, Y.-R.; Lo, Y.-T.; Lin, H.-Y.; Lin, S.-P. Genetic and Phenotypic Spectrum of KMT2D Variants in Taiwanese Case Series of Kabuki Syndrome. Diagnostics 2024, 14, 1815. https://doi.org/10.3390/diagnostics14161815
Lee C-L, Chuang C-K, Chen M-R, Lin J-L, Chiu H-C, Chang Y-H, Tu Y-R, Lo Y-T, Lin H-Y, Lin S-P. Genetic and Phenotypic Spectrum of KMT2D Variants in Taiwanese Case Series of Kabuki Syndrome. Diagnostics. 2024; 14(16):1815. https://doi.org/10.3390/diagnostics14161815
Chicago/Turabian StyleLee, Chung-Lin, Chih-Kuang Chuang, Ming-Ren Chen, Ju-Li Lin, Huei-Ching Chiu, Ya-Hui Chang, Yuan-Rong Tu, Yun-Ting Lo, Hsiang-Yu Lin, and Shuan-Pei Lin. 2024. "Genetic and Phenotypic Spectrum of KMT2D Variants in Taiwanese Case Series of Kabuki Syndrome" Diagnostics 14, no. 16: 1815. https://doi.org/10.3390/diagnostics14161815