
Anderson–Fabry Disease: Red Flags for Early Diagnosis of Cardiac Involvement
 , , ,
, , ,  , , , ,
, , , ,
Abstract
1. Introduction
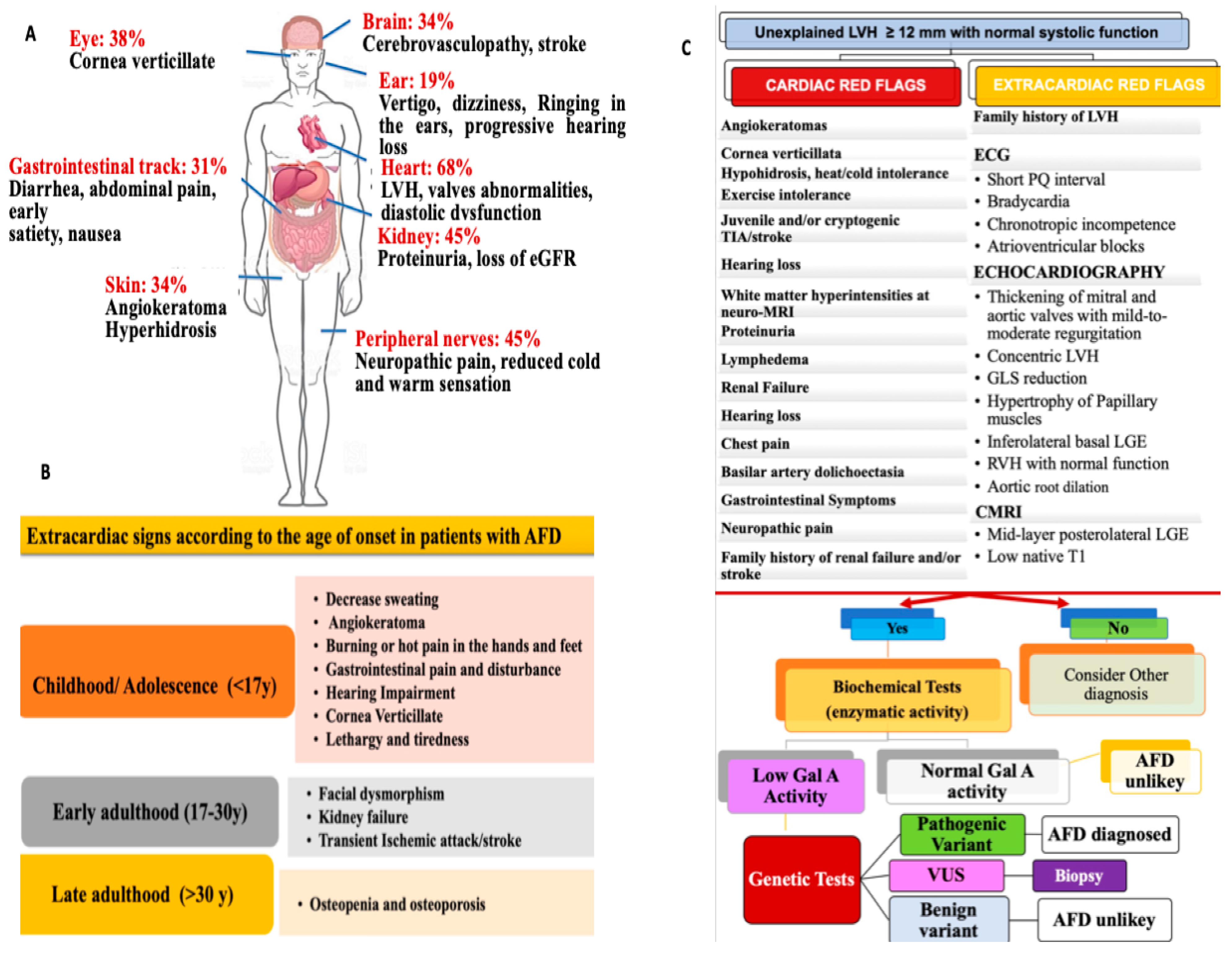
2. General Features and Clinical Presentation of AFD
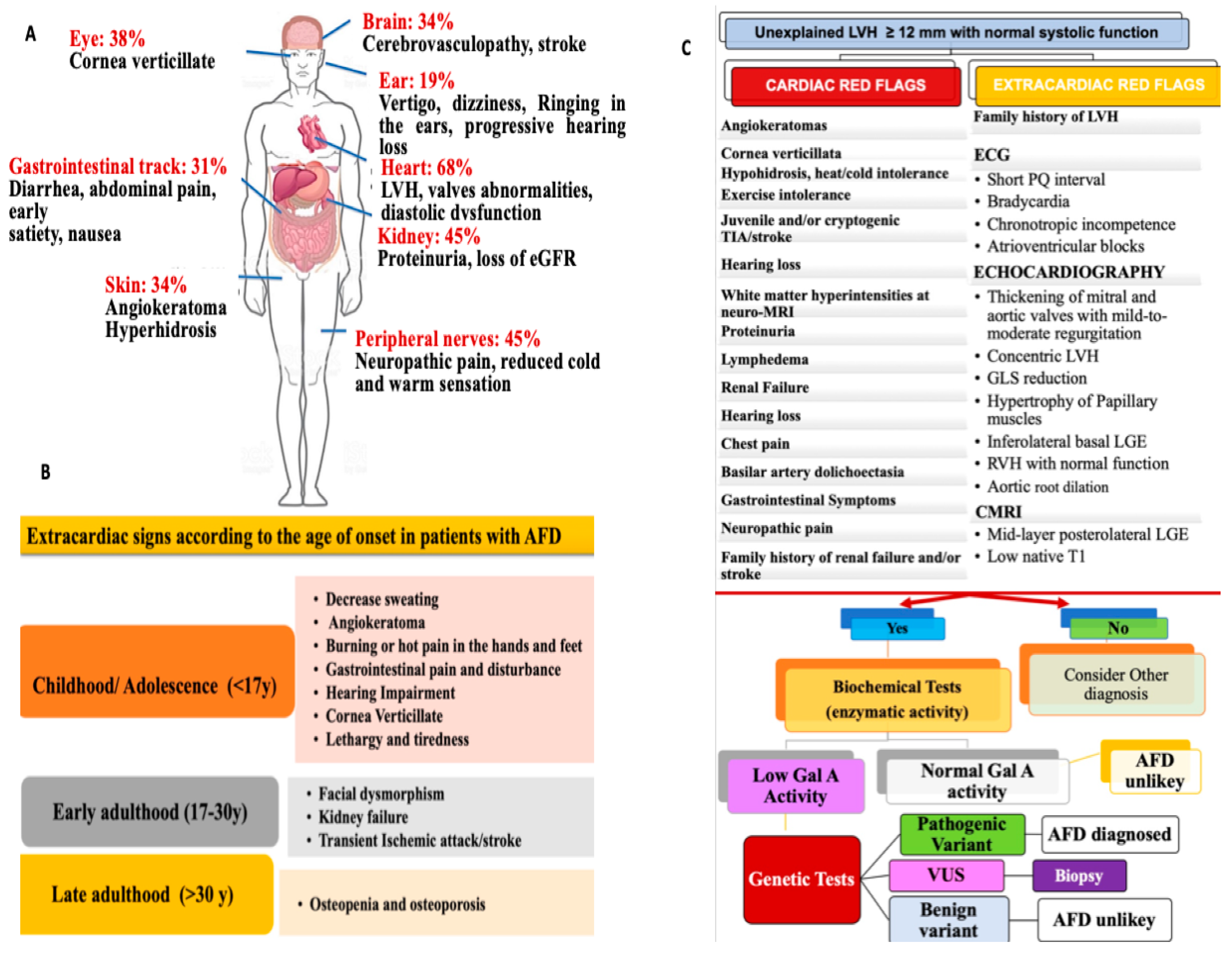
3. Cardiac Involvement
3.1. Pathophysiology
3.2. Disease Manifestations: Patient Symptoms
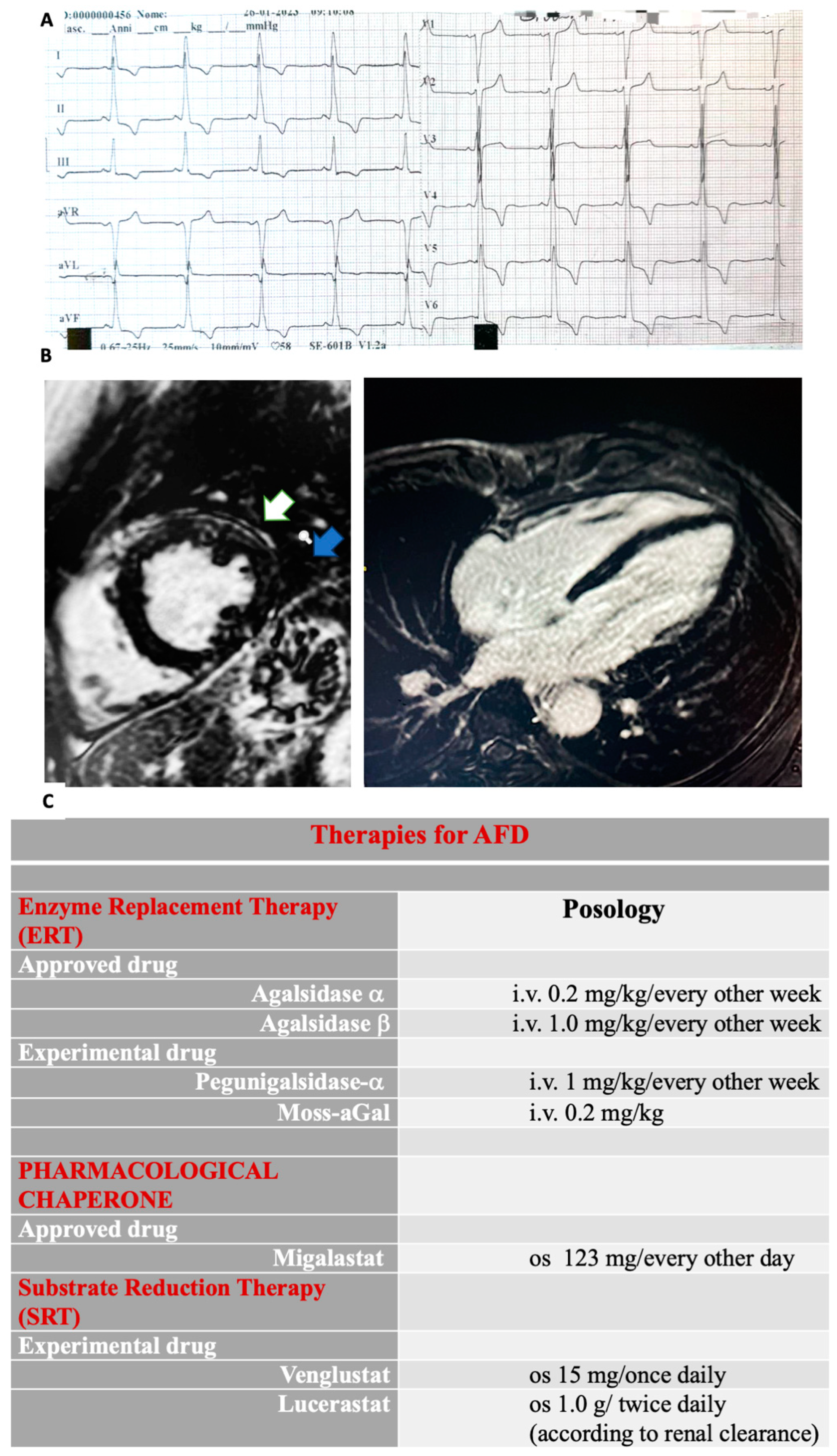
3.2.1. Electrophysiologic Abnormalities and Arrhythmias Burden
3.2.2. Echocardiographic Findings
3.2.3. Cardiac Magnetic Resonance Imaging Findings
4. Diagnostic Workup: The Roles of Genetic and Biochemical Testing, Biopsy, and Biomarkers in AFD
5. Therapy
6. Future Directions
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Marian, A.J. Challenges in the diagnosis of anderson-fabry disease. J. Am. Coll. Cardiol. 2016, 68, 1051–1053. [Google Scholar] [PubMed]
- Pieroni, M.; Moon, J.C.; Arbustini, E.; Barriales-Villa, R.; Camporeale, A.; Vujkovac, A.C.; Elliott, P.M.; Hagege, A.; Kuusisto, J.; Linhart, A.; et al. Cardiac involvement in fabry disease: Jacc review topic of the week. J. Am. Coll. Cardiol. 2021, 77, 922–936. [Google Scholar] [PubMed]
- Waldek, S.; Patel, M.R.; Banikazemi, M.; Lemay, R.; Lee, P. Life expectancy and cause of death in males and females with fabry disease: Findings from the fabry registry. Genet. Med. 2009, 11, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Vardarli, I.; Weber, M.; Rischpler, C.; Führer, D.; Herrmann, K.; Weidemann, F. Fabry cardiomyopathy: Current treatment and future options. J. Clin. Med. 2021, 10, 2750. [Google Scholar] [CrossRef]
- Ortiz, A.; Abiose, A.; Bichet, D.G.; Cabrera, G.; Charrow, J.; Germain, D.P.; Hopkin, R.J.; Jovanovic, A.; Linhart, A.; Maruti, S.S.; et al. Time to treatment benefit for adult patients with fabry disease receiving agalsidase β: Data from the fabry registry. J. Med. Genet. 2016, 53, 495–502. [Google Scholar]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Citro, R.; Prota, C.; Ferraioli, D.; Iuliano, G.; Bellino, M.; Radano, I.; Silverio, A.; Migliarino, S.; Polito, M.V.; Ruggiero, A.; et al. Importance of echocardiography and clinical “red flags” in guiding genetic screening for fabry disease. Front. Cardiovasc. Med. 2022, 9, 838200. [Google Scholar] [CrossRef]
- Baig, S.; Edward, N.C.; Kotecha, D.; Liu, B.; Nordin, S.; Kozor, R.; Moon, J.C.; Geberhiwot, T.; Steeds, R.P. Ventricular arrhythmia and sudden cardiac death in fabry disease: A systematic review of risk factors in clinical practice. Europace 2018, 20, f153–f161. [Google Scholar] [CrossRef]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. Jama 1999, 281, 249–254. [Google Scholar]
- Inoue, T.; Hattori, K.; Ihara, K.; Ishii, A.; Nakamura, K.; Hirose, S. Newborn screening for fabry disease in japan: Prevalence and genotypes of fabry disease in a pilot study. J. Hum. Genet. 2013, 58, 548–552. [Google Scholar]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Kittner, S.J.; Tuhrim, S.; Cole, J.W.; Stern, B.; Dobbins, M.; Grace, M.E.; Nazarenko, I.; Dobrovolny, R.; McDade, E.; et al. Frequency of unrecognized fabry disease among young european-american and african-american men with first ischemic stroke. Stroke 2010, 41, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Linhart, A.; Elliott, P.M. The heart in anderson-fabry disease and other lysosomal storage disorders. Heart 2007, 93, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Favalli, V.; Disabella, E.; Molinaro, M.; Tagliani, M.; Scarabotto, A.; Serio, A.; Grasso, M.; Narula, N.; Giorgianni, C.; Caspani, C.; et al. Genetic screening of anderson-fabry disease in probands referred from multispecialty clinics. J. Am. Coll. Cardiol. 2016, 68, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Zarate, Y.A.; Hopkin, R.J. Fabry’s disease. Lancet 2008, 372, 1427–1435. [Google Scholar] [PubMed]
- Mehta, A.; Clarke, J.T.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassmann, G. Natural course of fabry disease: Changing pattern of causes of death in fos—fabry outcome survey. J. Med. Genet. 2009, 46, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; O’Mahony, C.; Hughes, D.; Rahman, M.S.; Coats, C.; Murphy, E.; Lachmann, R.; Mehta, A.; Elliott, P.M. Clinical and genetic predictors of major cardiac events in patients with anderson-fabry disease. Heart 2015, 101, 961–966. [Google Scholar]
- Akhtar, M.M.; Elliott, P.M. Anderson-fabry disease in heart failure. Biophys. Rev. 2018, 10, 1107–1119. [Google Scholar] [CrossRef]
- Nakao, S.; Takenaka, T.; Maeda, M.; Kodama, C.; Tanaka, A.; Tahara, M.; Yoshida, A.; Kuriyama, M.; Hayashibe, H.; Sakuraba, H.; et al. An atypical variant of fabry’s disease in men with left ventricular hypertrophy. N. Engl. J. Med. 1995, 333, 288–293. [Google Scholar] [CrossRef]
- von Scheidt, W.; Eng, C.M.; Fitzmaurice, T.F.; Erdmann, E.; Hübner, G.; Olsen, E.G.; Christomanou, H.; Kandolf, R.; Bishop, D.F.; Desnick, R.J. An atypical variant of fabry’s disease with manifestations confined to the myocardium. N. Engl. J. Med. 1991, 324, 395–399. [Google Scholar] [CrossRef]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Ditaranto, R.; Graziani, F.; Tanini, I.; Camporeale, A.; Lillo, R.; Rubino, M.; Panaioli, E.; Di Nicola, F.; Ferrara, V.; et al. Standard ecg for differential diagnosis between anderson-fabry disease and hypertrophic cardiomyopathy. Heart 2022, 108, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Doheny, D.; Srinivasan, R.; Pagant, S.; Chen, B.; Yasuda, M.; Desnick, R.J. Fabry disease: Prevalence of affected males and heterozygotes with pathogenic gla mutations identified by screening renal, cardiac and stroke clinics, 1995–2017. J Med Genet 2018, 55, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Stankowski, K.; Figliozzi, S.; Battaglia, V.; Catapano, F.; Francone, M.; Monti, L. Fabry disease: More than a phenocopy of hypertrophic cardiomyopathy. J. Clin. Med. 2023, 12, 7061. [Google Scholar]
- Militaru, S.; Jurcuț, R.; Adam, R.; Roşca, M.; Ginghina, C.; Popescu, B.A. Echocardiographic features of fabry cardiomyopathy-comparison with hypertrophy-matched sarcomeric hypertrophic cardiomyopathy. Echocardiography 2019, 36, 2041–2049. [Google Scholar] [CrossRef]
- Karur, G.R.; Robison, S.; Iwanochko, R.M.; Morel, C.F.; Crean, A.M.; Thavendiranathan, P.; Nguyen, E.T.; Mathur, S.; Wasim, S.; Hanneman, K. Use of myocardial t1 mapping at 3.0 t to differentiate anderson-fabry disease from hypertrophic cardiomyopathy. Radiology 2018, 288, 398–406. [Google Scholar]
- Wechalekar, A.D.; Fontana, M.; Quarta, C.C.; Liedtke, M. Al amyloidosis for cardiologists: Awareness, diagnosis, and future prospects: Jacc: Cardiooncology state-of-the-art review. JACC CardioOncol 2022, 4, 427–441. [Google Scholar] [CrossRef]
- Azevedo, O.; Cordeiro, F.; Gago, M.F.; Miltenberger-Miltenyi, G.; Ferreira, C.; Sousa, N.; Cunha, D. Fabry disease and the heart: A comprehensive review. Int. J. Mol. Sci. 2021, 22, 4434. [Google Scholar]
- Frustaci, A.; Morgante, E.; Russo, M.A.; Scopelliti, F.; Grande, C.; Verardo, R.; Franciosa, P.; Chimenti, C. Pathology and function of conduction tissue in fabry disease cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2015, 8, 799–805. [Google Scholar] [CrossRef]
- Ikari, Y.; Kuwako, K.; Yamaguchi, T. Fabry’s disease with complete atrioventricular block: Histological evidence of involvement of the conduction system. Br. Heart J. 1992, 68, 323–325. [Google Scholar] [CrossRef]
- Mehta, A.; Ricci, R.; Widmer, U.; Dehout, F.; Garcia de Lorenzo, A.; Kampmann, C.; Linhart, A.; Sunder-Plassmann, G.; Ries, M.; Beck, M. Fabry disease defined: Baseline clinical manifestations of 366 patients in the fabry outcome survey. Eur. J. Clin. Invest. 2004, 34, 236–242. [Google Scholar]
- Linhart, A.; Kampmann, C.; Zamorano, J.L.; Sunder-Plassmann, G.; Beck, M.; Mehta, A.; Elliott, P.M. Cardiac manifestations of anderson-fabry disease: Results from the international fabry outcome survey. Eur. Heart J. 2007, 28, 1228–1235. [Google Scholar] [PubMed]
- Wu, J.C.; Ho, C.Y.; Skali, H.; Abichandani, R.; Wilcox, W.R.; Banikazemi, M.; Packman, S.; Sims, K.; Solomon, S.D. Cardiovascular manifestations of fabry disease: Relationships between left ventricular hypertrophy, disease severity, and alpha-galactosidase a activity. Eur. Heart J. 2010, 31, 1088–1097. [Google Scholar] [PubMed]
- Selthofer-Relatic, K. Time of anderson-fabry disease detection and cardiovascular presentation. Case Rep. Cardiol. 2018, 2018, 6131083. [Google Scholar] [PubMed]
- Clarke, J.T.; Giugliani, R.; Sunder-Plassmann, G.; Elliott, P.M.; Pintos-Morell, G.; Hernberg-Ståhl, E.; Malmenäs, M.; Beck, M. Impact of measures to enhance the value of observational surveys in rare diseases: The fabry outcome survey (fos). Value Health 2011, 14, 862–866. [Google Scholar]
- Eng, C.M.; Fletcher, J.; Wilcox, W.R.; Waldek, S.; Scott, C.R.; Sillence, D.O.; Breunig, F.; Charrow, J.; Germain, D.P.; Nicholls, K.; et al. Fabry disease: Baseline medical characteristics of a cohort of 1765 males and females in the fabry registry. J. Inherit. Metab. Dis. 2007, 30, 184–192. [Google Scholar]
- Pieroni, M.; Chimenti, C.; Ricci, R.; Sale, P.; Russo, M.A.; Frustaci, A. Early detection of fabry cardiomyopathy by tissue doppler imaging. Circulation 2003, 107, 1978–1984. [Google Scholar] [CrossRef]
- Putko, B.N.; Wen, K.; Thompson, R.B.; Mullen, J.; Shanks, M.; Yogasundaram, H.; Sergi, C.; Oudit, G.Y. Anderson-fabry cardiomyopathy: Prevalence, pathophysiology, diagnosis and treatment. Heart Fail. Rev. 2015, 20, 179–191. [Google Scholar]
- Chimenti, C.; Morgante, E.; Tanzilli, G.; Mangieri, E.; Critelli, G.; Gaudio, C.; Russo, M.A.; Frustaci, A. Angina in fabry disease reflects coronary small vessel disease. Circ. Heart Fail. 2008, 1, 161–169. [Google Scholar] [CrossRef]
- Elleder, M. Sequelae of storage in fabry disease—pathology and comparison with other lysosomal storage diseases. Acta Paediatr. Suppl. 2003, 92, 46–53; discussion 45. [Google Scholar] [CrossRef]
- Chimenti, C.; Morgante, E.; Critelli, G.; Russo, M.A.; Frustaci, A. Coronary artery bypass grafting for fabry’s disease: Veins more suitable than arteries? Hum. Pathol. 2007, 38, 1864–1867. [Google Scholar] [PubMed]
- Namdar, M. Electrocardiographic changes and arrhythmia in fabry disease. Front. Cardiovasc. Med. 2016, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, C.; Wiethoff, C.M.; Whybra, C.; Baehner, F.A.; Mengel, E.; Beck, M. Cardiac manifestations of anderson-fabry disease in children and adolescents. Acta Paediatr. 2008, 97, 463–469. [Google Scholar] [CrossRef]
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.; Charron, P.; Gimeno-Blanes, J.; Heliö, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the esc working group on myocardial and pericardial diseases. Eur. Heart J. 2013, 34, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Namdar, M.; Steffel, J.; Vidovic, M.; Brunckhorst, C.B.; Holzmeister, J.; Lüscher, T.F.; Jenni, R.; Duru, F. Electrocardiographic changes in early recognition of fabry disease. Heart 2011, 97, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Yousef, Z.; Elliott, P.M.; Cecchi, F.; Escoubet, B.; Linhart, A.; Monserrat, L.; Namdar, M.; Weidemann, F. Left ventricular hypertrophy in fabry disease: A practical approach to diagnosis. Eur. Heart J. 2013, 34, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, C.; Russo, M.A.; Frustaci, A. Atrial biopsy evidence of fabry disease causing lone atrial fibrillation. Heart 2010, 96, 1782–1783. [Google Scholar]
- Shah, J.S.; Hughes, D.A.; Sachdev, B.; Tome, M.; Ward, D.; Lee, P.; Mehta, A.B.; Elliott, P.M. Prevalence and clinical significance of cardiac arrhythmia in anderson-fabry disease. Am. J. Cardiol. 2005, 96, 842–846. [Google Scholar] [CrossRef]
- Yeung, D.F.; Sirrs, S.; Tsang, M.Y.C.; Gin, K.; Luong, C.; Jue, J.; Nair, P.; Lee, P.K.; Tsang, T.S.M. Echocardiographic assessment of patients with fabry disease. J. Am. Soc. Echocardiogr. 2018, 31, 639–649.e632. [Google Scholar]
- Tower-Rader, A.; Jaber, W.A. Multimodality imaging assessment of fabry disease. Circ. Cardiovasc. Imaging 2019, 12, e009013. [Google Scholar]
- Linhart, A.; Palecek, T.; Bultas, J.; Ferguson, J.J.; Hrudová, J.; Karetová, D.; Zeman, J.; Ledvinová, J.; Poupetová, H.; Elleder, M.; et al. New insights in cardiac structural changes in patients with fabry’s disease. Am. Heart J. 2000, 139, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, B.; Takenaka, T.; Teraguchi, H.; Tei, C.; Lee, P.; McKenna, W.J.; Elliott, P.M. Prevalence of anderson-fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002, 105, 1407–1411. [Google Scholar]
- Calcagnino, M.; O’Mahony, C.; Coats, C.; Cardona, M.; Garcia, A.; Janagarajan, K.; Mehta, A.; Hughes, D.; Murphy, E.; Lachmann, R.; et al. Exercise-induced left ventricular outflow tract obstruction in symptomatic patients with anderson-fabry disease. J. Am. Coll. Cardiol. 2011, 58, 88–89. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Beck, M.; Sunder-Plassmann, G. Fabry Disease: Perspectives from 5 Years of FOS; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Chimenti, C.; Ricci, R.; Pieroni, M.; Natale, L.; Russo, M.A.; Frustaci, A. Cardiac variant of fabry’s disease mimicking hypertrophic cardiomyopathy. Cardiologia 1999, 44, 469–473. [Google Scholar]
- Weidemann, F.; Breunig, F.; Beer, M.; Sandstede, J.; Störk, S.; Voelker, W.; Ertl, G.; Knoll, A.; Wanner, C.; Strotmann, J.M. The variation of morphological and functional cardiac manifestation in fabry disease: Potential implications for the time course of the disease. Eur. Heart J. 2005, 26, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Labombarda, F.; Saloux, E.; Milesi, G.; Bienvenu, B. Loss of base-to-apex circumferential strain gradient: A specific pattern of fabry cardiomyopathy? Echocardiography 2017, 34, 504–510. [Google Scholar] [PubMed]
- Shah, J.S.; Lee, P.; Hughes, D.; Thaman, R.; Sachdev, B.; Pellerin, D.; Mehta, A.; Elliott, P.M. The natural history of left ventricular systolic function in anderson-fabry disease. Heart 2005, 91, 533–534. [Google Scholar] [CrossRef]
- Spinelli, L.; Giugliano, G.; Pisani, A.; Imbriaco, M.; Riccio, E.; Russo, C.; Cuocolo, A.; Trimarco, B.; Esposito, G. Does left ventricular function predict cardiac outcome in anderson-fabry disease? Int. J. Cardiovasc. Imaging 2021, 37, 1225–1236. [Google Scholar]
- Takenaka, T.; Teraguchi, H.; Yoshida, A.; Taguchi, S.; Ninomiya, K.; Umekita, Y.; Yoshida, H.; Horinouchi, M.; Tabata, K.; Yonezawa, S.; et al. Terminal stage cardiac findings in patients with cardiac fabry disease: An electrocardiographic, echocardiographic, and autopsy study. J. Cardiol. 2008, 51, 50–59. [Google Scholar]
- Deva, D.P.; Hanneman, K.; Li, Q.; Ng, M.Y.; Wasim, S.; Morel, C.; Iwanochko, R.M.; Thavendiranathan, P.; Crean, A.M. Cardiovascular magnetic resonance demonstration of the spectrum of morphological phenotypes and patterns of myocardial scarring in anderson-fabry disease. J. Cardiovasc. Magn. Reson. 2016, 18, 14. [Google Scholar] [CrossRef]
- Weidemann, F.; Sommer, C.; Duning, T.; Lanzl, I.; Möhrenschlager, M.; Naleschinski, D.; Arning, K.; Baron, R.; Niemann, M.; Breunig, F.; et al. Department-related tasks and organ-targeted therapy in fabry disease: An interdisciplinary challenge. Am. J. Med. 2010, 123, e651–e658. [Google Scholar] [CrossRef] [PubMed]
- Messroghli, D.R.; Moon, J.C.; Ferreira, V.M.; Grosse-Wortmann, L.; He, T.; Kellman, P.; Mascherbauer, J.; Nezafat, R.; Salerno, M.; Schelbert, E.B.; et al. Clinical recommendations for cardiovascular magnetic resonance mapping of t1, t2, t2* and extracellular volume: A consensus statement by the society for cardiovascular magnetic resonance (SCMR) endorsed by the european association for cardiovascular imaging (EACVI). J. Cardiovasc. Magn. Reson. 2017, 19, 75. [Google Scholar] [PubMed]
- Schelbert, E.B.; Messroghli, D.R. State of the art: Clinical applications of cardiac t1 mapping. Radiology 2016, 278, 658–676. [Google Scholar] [PubMed]
- Augusto, J.B.; Johner, N.; Shah, D.; Nordin, S.; Knott, K.D.; Rosmini, S.; Lau, C.; Alfarih, M.; Hughes, R.; Seraphim, A.; et al. The myocardial phenotype of fabry disease pre-hypertrophy and pre-detectable storage. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 790–799. [Google Scholar] [CrossRef]
- Ho, C.Y.; Abbasi, S.A.; Neilan, T.G.; Shah, R.V.; Chen, Y.; Heydari, B.; Cirino, A.L.; Lakdawala, N.K.; Orav, E.J.; González, A.; et al. T1 measurements identify extracellular volume expansion in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. Circ. Cardiovasc. Imaging 2013, 6, 415–422. [Google Scholar]
- Sado, D.M.; White, S.K.; Piechnik, S.K.; Banypersad, S.M.; Treibel, T.; Captur, G.; Fontana, M.; Maestrini, V.; Flett, A.S.; Robson, M.D.; et al. Identification and assessment of anderson-fabry disease by cardiovascular magnetic resonance noncontrast myocardial t1 mapping. Circ. Cardiovasc. Imaging 2013, 6, 392–398. [Google Scholar] [CrossRef]
- Pica, S.; Sado, D.M.; Maestrini, V.; Fontana, M.; White, S.K.; Treibel, T.; Captur, G.; Anderson, S.; Piechnik, S.K.; Robson, M.D.; et al. Reproducibility of native myocardial t1 mapping in the assessment of fabry disease and its role in early detection of cardiac involvement by cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2014, 16, 99. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 esc guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef]
- Nordin, S.; Kozor, R.; Medina-Menacho, K.; Abdel-Gadir, A.; Baig, S.; Sado, D.M.; Lobascio, I.; Murphy, E.; Lachmann, R.H.; Mehta, A.; et al. Proposed stages of myocardial phenotype development in fabry disease. JACC Cardiovasc. Imaging 2019, 12, 1673–1683. [Google Scholar] [CrossRef]
- Thompson, R.B.; Chow, K.; Khan, A.; Chan, A.; Shanks, M.; Paterson, I.; Oudit, G.Y. T1 mapping with cardiovascular mri is highly sensitive for fabry disease independent of hypertrophy and sex. Circ. Cardiovasc. Imaging 2013, 6, 637–645. [Google Scholar] [CrossRef]
- Linthorst, G.E.; Vedder, A.C.; Aerts, J.M.; Hollak, C.E. Screening for fabry disease using whole blood spots fails to identify one-third of female carriers. Clin. Chim. Acta 2005, 353, 201–203. [Google Scholar] [CrossRef] [PubMed]
- D Valle, B.V.; Kinzel, K.W.; Antonarakis, S.E.; Ballabio, A.; Gibson, K.M.; Mithcell, G. The Metabolic e Molecular Bases of Inheredited Disease, 8th ed.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Smid, B.E.; van der Tol, L.; Cecchi, F.; Elliott, P.M.; Hughes, D.A.; Linthorst, G.E.; Timmermans, J.; Weidemann, F.; West, M.L.; Biegstraaten, M.; et al. Uncertain diagnosis of fabry disease: Consensus recommendation on diagnosis in adults with left ventricular hypertrophy and genetic variants of unknown significance. Int. J. Cardiol. 2014, 177, 400–408. [Google Scholar] [CrossRef]
- van der Tol, L.; Cassiman, D.; Houge, G.; Janssen, M.C.; Lachmann, R.H.; Linthorst, G.E.; Ramaswami, U.; Sommer, C.; Tøndel, C.; West, M.L.; et al. Uncertain diagnosis of fabry disease in patients with neuropathic pain, angiokeratoma or cornea verticillata: Consensus on the approach to diagnosis and follow-up. JIMD Rep. 2014, 17, 83–90. [Google Scholar] [PubMed]
- Politei, J.; Frabasil, J.; Durand, C.; Di Pietrantonio, S.; Fernandez, A.; Albertón, V.; Velasquez Rivas, D.; Barriales-Villa, R.; Larrañaga-Moreira, J.; Schenone, A.B. Incidental finding of cornea verticillata or lamellar inclusions in kidney biopsy: Measurement of lyso-gb3 in plasma defines between fabry disease and drug-induced phospholipidosis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 165985. [Google Scholar] [CrossRef] [PubMed]
- Bracamonte, E.R.; Kowalewska, J.; Starr, J.; Gitomer, J.; Alpers, C.E. Iatrogenic phospholipidosis mimicking fabry disease. Am. J. Kidney Dis. 2006, 48, 844–850. [Google Scholar]
- Maruyama, H.; Miyata, K.; Mikame, M.; Taguchi, A.; Guili, C.; Shimura, M.; Murayama, K.; Inoue, T.; Yamamoto, S.; Sugimura, K.; et al. Effectiveness of plasma lyso-gb3 as a biomarker for selecting high-risk patients with fabry disease from multispecialty clinics for genetic analysis. Genet. Med. 2019, 21, 44–52. [Google Scholar]
- Togawa, T.; Kodama, T.; Suzuki, T.; Sugawara, K.; Tsukimura, T.; Ohashi, T.; Ishige, N.; Suzuki, K.; Kitagawa, T.; Sakuraba, H. Plasma globotriaosylsphingosine as a biomarker of fabry disease. Mol. Genet. Metab. 2010, 100, 257–261. [Google Scholar] [CrossRef]
- Seydelmann, N.; Liu, D.; Krämer, J.; Drechsler, C.; Hu, K.; Nordbeck, P.; Schneider, A.; Störk, S.; Bijnens, B.; Ertl, G.; et al. High-sensitivity troponin: A clinical blood biomarker for staging cardiomyopathy in fabry disease. J. Am. Heart Assoc. 2016, 5, e002839. [Google Scholar] [CrossRef]
- Coats, C.J.; Parisi, V.; Ramos, M.; Janagarajan, K.; O’Mahony, C.; Dawnay, A.; Lachmann, R.H.; Murphy, E.; Mehta, A.; Hughes, D.; et al. Role of serum n-terminal pro-brain natriuretic peptide measurement in diagnosis of cardiac involvement in patients with anderson-fabry disease. Am. J. Cardiol. 2013, 111, 111–117. [Google Scholar]
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. Safety and efficacy of recombinant human alpha-galactosidase a replacement therapy in fabry’s disease. N. Engl. J. Med. 2001, 345, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Kopp, J.B.; Austin, H.A., 3rd; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme replacement therapy in fabry disease: A randomized controlled trial. Jama 2001, 285, 2743–2749. [Google Scholar] [CrossRef]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of fabry’s disease with the pharmacologic chaperone migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar]
- Benjamin, E.R.; Della Valle, M.C.; Wu, X.; Katz, E.; Pruthi, F.; Bond, S.; Bronfin, B.; Williams, H.; Yu, J.; Bichet, D.G.; et al. The validation of pharmacogenetics for the identification of fabry patients to be treated with migalastat. Genet. Med. 2017, 19, 430–438. [Google Scholar]
- Schiffmann, R.; Murray, G.J.; Treco, D.; Daniel, P.; Sellos-Moura, M.; Myers, M.; Quirk, J.M.; Zirzow, G.C.; Borowski, M.; Loveday, K.; et al. Infusion of alpha-galactosidase a reduces tissue globotriaosylceramide storage in patients with fabry disease. Proc. Natl. Acad. Sci. USA 2000, 97, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, L.; Pisani, A.; Sabbatini, M.; Petretta, M.; Andreucci, M.V.; Procaccini, D.; Lo Surdo, N.; Federico, S.; Cianciaruso, B. Enzyme replacement therapy with agalsidase beta improves cardiac involvement in fabry’s disease. Clin. Genet. 2004, 66, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Arends, M.; Biegstraaten, M.; Wanner, C.; Sirrs, S.; Mehta, A.; Elliott, P.M.; Oder, D.; Watkinson, O.T.; Bichet, D.G.; Khan, A.; et al. Agalsidase alfa versus agalsidase beta for the treatment of fabry disease: An international cohort study. J. Med. Genet. 2018, 55, 351–358. [Google Scholar] [CrossRef]
- Weidemann, F.; Niemann, M.; Störk, S.; Breunig, F.; Beer, M.; Sommer, C.; Herrmann, S.; Ertl, G.; Wanner, C. Long-term outcome of enzyme-replacement therapy in advanced fabry disease: Evidence for disease progression towards serious complications. J. Intern. Med. 2013, 274, 331–341. [Google Scholar] [CrossRef]
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagège, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of fabry disease. Eur. J. Heart Fail. 2020, 22, 1076–1096. [Google Scholar]
- Germain, D.P.; Elliott, P.M.; Falissard, B.; Fomin, V.V.; Hilz, M.J.; Jovanovic, A.; Kantola, I.; Linhart, A.; Mignani, R.; Namdar, M.; et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with fabry disease: A systematic literature review by a european panel of experts. Mol. Genet. Metab. Rep. 2019, 19, 100454. [Google Scholar] [CrossRef]
- Weidemann, F.; Niemann, M.; Breunig, F.; Herrmann, S.; Beer, M.; Störk, S.; Voelker, W.; Ertl, G.; Wanner, C.; Strotmann, J. Long-term effects of enzyme replacement therapy on fabry cardiomyopathy: Evidence for a better outcome with early treatment. Circulation 2009, 119, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Weidemann, F.; Abiose, A.; Patel, M.R.; Cizmarik, M.; Cole, J.A.; Beitner-Johnson, D.; Benistan, K.; Cabrera, G.; Charrow, J.; et al. Analysis of left ventricular mass in untreated men and in men treated with agalsidase-β: Data from the fabry registry. Genet. Med. 2013, 15, 958–965. [Google Scholar] [CrossRef] [PubMed]
- El Dib, R.; Gomaa, H.; Ortiz, A.; Politei, J.; Kapoor, A.; Barreto, F. Enzyme replacement therapy for anderson-fabry disease: A complementary overview of a cochrane publication through a linear regression and a pooled analysis of proportions from cohort studies. PLoS ONE 2017, 12, e0173358. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Breunig, F.; Beer, M.; Sandstede, J.; Turschner, O.; Voelker, W.; Ertl, G.; Knoll, A.; Wanner, C.; Strotmann, J.M. Improvement of cardiac function during enzyme replacement therapy in patients with fabry disease: A prospective strain rate imaging study. Circulation 2003, 108, 1299–1301. [Google Scholar] [PubMed]
- Parenti, G.; Moracci, M.; Fecarotta, S.; Andria, G. Pharmacological chaperone therapy for lysosomal storage diseases. Future Med. Chem. 2014, 6, 1031–1045. [Google Scholar] [PubMed]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in fabry disease: 18-month results from the randomised phase iii attract study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef]
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Eveslage, M.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Müntze, J.; et al. Treatment of fabry disease management with migalastat-outcome from a prospective 24 months observational multicenter study (famous). Eur. Heart J. Cardiovasc. Pharmacother. 2022, 8, 272–281. [Google Scholar]
- Narita, I.; Ohashi, T.; Sakai, N.; Hamazaki, T.; Skuban, N.; Castelli, J.P.; Lagast, H.; Barth, J.A. Efficacy and safety of migalastat in a japanese population: A subgroup analysis of the attract study. Clin. Exp. Nephrol. 2020, 24, 157–166. [Google Scholar]
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Müntze, J.; Üçeyler, N.; et al. Treatment of fabry’s disease with migalastat: Outcome from a prospective observational multicenter study (famous). Clin. Pharmacol. Ther. 2020, 108, 326–337. [Google Scholar]
- Feldt-Rasmussen, U.; Hughes, D.; Sunder-Plassmann, G.; Shankar, S.; Nedd, K.; Olivotto, I.; Ortiz, D.; Ohashi, T.; Hamazaki, T.; Skuban, N.; et al. Long-term efficacy and safety of migalastat treatment in fabry disease: 30-month results from the open-label extension of the randomized, phase 3 attract study. Mol. Genet. Metab. 2020, 131, 219–228. [Google Scholar]
- Germain, D.P.; Nicholls, K.; Giugliani, R.; Bichet, D.G.; Hughes, D.A.; Barisoni, L.M.; Colvin, R.B.; Jennette, J.C.; Skuban, N.; Castelli, J.P.; et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of fabry disease and migalastat-amenable variants: Data from the phase 3 randomized, multicenter, double-blind clinical trial and extension study. Genet. Med. 2019, 21, 1987–1997. [Google Scholar] [PubMed]
- Baehner, F.; Kampmann, C.; Whybra, C.; Miebach, E.; Wiethoff, C.M.; Beck, M. Enzyme replacement therapy in heterozygous females with fabry disease: Results of a phase iiib study. J. Inherit. Metab. Dis. 2003, 26, 617–627. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
| AFD | CA | HCM | |
|---|---|---|---|
| ECG |
|
|
|
| Biochemical |
|
|
|
| Clinical |
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iorio, A.; Lucà, F.; Pozzi, A.; Rao, C.M.; Chimenti, C.; Di Fusco, S.A.; Rossini, R.; Caretta, G.; Cornara, S.; Giubilato, S.; et al. Anderson–Fabry Disease: Red Flags for Early Diagnosis of Cardiac Involvement. Diagnostics 2024, 14, 208. https://doi.org/10.3390/diagnostics14020208
Iorio A, Lucà F, Pozzi A, Rao CM, Chimenti C, Di Fusco SA, Rossini R, Caretta G, Cornara S, Giubilato S, et al. Anderson–Fabry Disease: Red Flags for Early Diagnosis of Cardiac Involvement. Diagnostics. 2024; 14(2):208. https://doi.org/10.3390/diagnostics14020208
Chicago/Turabian StyleIorio, Annamaria, Fabiana Lucà, Andrea Pozzi, Carmelo Massimiliano Rao, Cristina Chimenti, Stefania Angela Di Fusco, Roberta Rossini, Giorgio Caretta, Stefano Cornara, Simona Giubilato, and et al. 2024. "Anderson–Fabry Disease: Red Flags for Early Diagnosis of Cardiac Involvement" Diagnostics 14, no. 2: 208. https://doi.org/10.3390/diagnostics14020208
APA StyleIorio, A., Lucà, F., Pozzi, A., Rao, C. M., Chimenti, C., Di Fusco, S. A., Rossini, R., Caretta, G., Cornara, S., Giubilato, S., Di Matteo, I., Di Nora, C., Pilleri, A., Gelsomino, S., Ceravolo, R., Riccio, C., Grimaldi, M., Colivicchi, F., Oliva, F., ... the Cardiac Rare Diseases Working Group Associazione Nazionale Medici Cardiologi Ospedalieri (ANMCO). (2024). Anderson–Fabry Disease: Red Flags for Early Diagnosis of Cardiac Involvement. Diagnostics, 14(2), 208. https://doi.org/10.3390/diagnostics14020208