Simple Summary
Anopheles gambiae is a major malaria vector. This vector is controlled mainly using insecticide, thereby reducing malaria transmission. Since many insecticides are toxic to non-target species, and there is increasing insecticide resistance, there is a need to identify other safer molecules that can be used to control the vector. Molecules inhibiting trehalase have been proposed as safer options for insecticide and fungicide development since humans do not produce trehalose. This work screens several trehalase inhibitors against trehalase of Anopheles gambiae (AgTre) and assesses their safety in humans using in silico methods. Four trehalase inhibitors had a high affinity for AgTre and were predicted to be relatively safe. The compounds also interacted well with trehalase of Aedes aegypti, another important mosquito vector. The results suggest these molecules are safer options for insecticide development to control mosquito-borne diseases.
Abstract
Trehalase inhibitors are considered safe alternatives for insecticides and fungicides. However, there are no studies testing these compounds on Anopheles gambiae, a major vector of human malaria. This study predicted the three-dimensional structure of Anopheles gambiae trehalase (AgTre) and identified potential inhibitors using molecular docking and molecular dynamics methods. Robetta server, C-I-TASSER, and I-TASSER were used to predict the protein structure, while the structural assessment was carried out using SWISS-MODEL, ERRAT, and VERIFY3D. Molecular docking and screening of 3022 compounds was carried out using AutoDock Vina in PyRx, and MD simulation was carried out using NAMD. The Robetta model outperformed all other models and was used for docking and simulation studies. After a post-screening analysis and ADMET studies, uniflorine, 67837201, 10406567, and Compound 2 were considered the best hits with binding energies of −6.9, −8.9, −9, and −8.4 kcal/mol, respectively, better than validamycin A standard (−5.4 kcal/mol). These four compounds were predicted to have no eco-toxicity, Brenk, or PAINS alerts. Similarly, they were predicted to be non-mutagenic, carcinogenic, or hepatoxic. 67837201, 10406567, and Compound 2 showed excellent stability during simulation. The study highlights uniflorine, 67837201, 10406567, and Compound 2 as good inhibitors of AgTre and possible compounds for malaria vector control.
Keywords:
trehalase; trehazolin; mosquito; vector control; validamycin; validoxylamine; uniflorine; casuarine 1. Introduction
Mosquitoes such as Anopheles and Aedes spp. are important vectors of several human diseases [1]. These mosquito-borne diseases, such as malaria, are controlled principally by employing vector-control strategies [2]. These strategies aim to reduce or eliminate the contact of the vector with humans, thus preventing parasite transmission. The primary vector control strategy for malaria involves the use of insecticides either in insecticide-treated nets (ITNs) or for indoor residual spraying (IRS) [3]. These strategies have contributed considerably to averting clinical cases of the disease, with about sixty-eight per cent of the 663 million averted cases between 2000 and 2015 attributed to the use of ITNs [4]. This highlights the importance of vector control in curbing malaria. However, increasing reports of insecticide resistance, the toxicity of insecticides to non-target species, and the persistence of insecticides in the environment have necessitated the search for safer alternatives in vector control [5,6]. Larval source management (LSM) strategies, which prevent the emergence of adult mosquitoes, are one of the complementary strategies employed in vector control [7]. These methods reduce the number of house-entering as well as outdoor biting mosquitoes and have been observed to reduce malaria transmission when combined with the use of ITNs [8]. Amidst this, increasing research efforts are being made to identify and develop new classes of safer compounds that could be used in vector control.
Trehalase inhibitors have been proposed as safer alternatives for insecticides as they are non-toxic and do not persist in the environment [9]. Trehalase (EC 3.2.1.28) is an enzyme that catalyses the reduction of trehalose into two molecules of glucose [10,11]. This reaction is critical in insects, as trehalose is the major hemolymph sugar in insects and is also the fuel required for flight [12]. Trehalose is also needed in larvae to resist stress factors [13]. Trehalase levels have been reported to be upregulated in the salivary gland of Anopheles stephensi during Plasmodium vivax infection, suggesting that it might also play a crucial role in parasite development in the mosquito [14]. Similarly, inhibition of trehalose transporter in Anopheles gambiae was observed to lead to decreased survival of the mosquito as well as decreased Plasmodium development in the mosquito [15]. Inhibition of trehalase by validamycin has been proposed as promising for the development of new insecticides [16]. Validamycin A has been tested in different insect species. For example, it has been observed to cause developmental defects and decrease fecundity in Helicoverpa armigera [17] and cause mortality in Diaphorina citri [18]. Marten et al. [19] reported that validamycin A, a trehalase inhibitor, reduced egg hatching and pupation, skewed the sex ratio, and prevented flight in Aedes aegypti, the vector for dengue fever, chikungunya, Zika fever, Mayaro, and yellow fever. Minimal larva death in Ae. aegypti (7% mortality at 100 and 200 ppm, and 9% mortality at 500 ppm validamycin A administration) was observed in their study. An earlier report by Logan [20] revealed that validoxylamine A, another trehalase inhibitor, did not cause significant mortality of Ae. Aegypti larvae but prevented flight of emerging adults and decreased the number of eggs laid by adult Ae. aegypti mosquitoes. This suggests that trehalase inhibitors can serve as insect growth regulators, sterilants, and flight inhibitors. This is important as the flight of disease vectors to access humans is required for disease transmission. Similarly, sterilants can help to reduce mosquito populations. There is growing research on synthesising other potential trehalase inhibitors and testing their activities in different species as fungicides or insecticides [9]. Although these potential inhibitors have been tested in different organisms, no study has tested these compounds in the malaria vector, Anopheles spp., to elucidate their possible use for malaria vector control. While validamycin A and validoxylamine A have been tested in Ae. aegypti, validamycin A is considered a weaker binder of trehalase, and the safety of validoxylamine A is of concern, since it is a good binder of porcine trehalase. Hence, there is a need to identify more potent and safe inhibitors of trehalase aside from validamycin A in these disease vectors, which can be developed for vector control. These can be carried out using traditional drug design methods or computer-aided drug design methods (CADD).
Traditional drug design methods are costly as they involve the synthesis of numerous compounds followed by in vitro or in vivo testing of their activities. For example, D’Adamio et al. [13] synthesised more than 20 compounds and tested their activity against Chironomus riparius trehalase. Apart from the cost involved in synthesising numerous compounds, in vitro or in vivo testing of compounds depend on the availability of recombinant enzymes or the organisms, which come with a cost that multiplies with an increasing number of compounds. However, CADD methods, which include molecular docking and molecular dynamics (MD) simulation, offer cheaper and faster means of screening and identifying potential binders of a protein from a wide library of compounds [21]. These methods rely on the availability of the three-dimensional (3D) structure of the protein of interest and a compound library [22]. Protein structure can be experimentally determined or computationally derived [23]. Potential hits, obtained based on docking scores from molecular docking, can be optimised and re-screened, leading to the guided synthesis of compounds. This helps to reduce the costs involved in insecticide/drug development. Performing molecular dynamics simulation after molecular docking helps to reveal the stability of protein–ligand interactions and may separate false positive from true positive hits [21]. As described above, trehalase inhibitors are promising vector-control compounds. Unfortunately, there is no report about their use against An. gambiae, an important malaria vector. In this study, the 3D-structure of An. gambiae trehalase (AgTre) was determined and molecular docking was employed to screen a library of compounds in order to identify potential lead structures against AgTre that could be developed for malaria vector control. Considerations were given to the interaction of these compounds to trehalase of Ae. aegypti, which is also an important mosquito vector of human diseases. Since trehalase from both organisms share >70% sequence identity, inhibitors could be used to control both vectors, preventing transmission of other mosquito-borne diseases aside from malaria, e.g., dengue fever. In addition, the medicinal chemistry and toxicity potential of these compounds were predicted, and the stability of their complexes with trehalase was determined. Four trehalase inhibitors were observed to be better binders of trehalase of An. gambiae and Ae. aegypti than validamycin A. The compounds were predicted to be non-toxic when compared to validoxylamine. Hence, the study proposes these inhibitors as potential molecules for vector control.
2. Materials and Methods
2.1. Prediction of 3D Structure of AgTre
Most insects have at least two copies of trehalase genes; however, it has been reported that dipteran insects, such as Anopheles spp., have only one copy of trehalase [24]. The single copy gene of trehalase in An. gambiae is AGAP012053, which codes for its trehalase protein AgTre. The experimental 3D structure of AgTre has not been determined; hence it is not available in the database of the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) [25]. However, a predicted structure based on homology modelling is available in the SWISS-MODEL Repository [26]. The structure was built using trehalase of Arabidopsis thaliana (7E9X_A) as a template which has the highest percentage identity (33%) to AgTre. Considering the low percentage identity and family distance of the two proteins (plant to insect), ab initio modelling was implemented in predicting the 3D structure of AgTre. The FastA file containing the protein sequence of the AgTre (Trehalase from An. gambiae Pest strain) was retrieved from UniProt Knowledgebase (UniProtKB) [27] with the accession number Q7PZS4. The protein sequence of AgTre was submitted to three ab initio modelling web servers: Iterative Threading Assembly Refinement (I-TASSER), Contact-guided Iterative Threading ASSEmbly Refinement (C-I-TASSER) [28,29], and the Robetta web server. On the Robetta server, a deep learning-based modelling method, RoseTTAFold [30], was used to predict the 3D structure of AgTre. RoseTTAFold gives confidence scores for models (ranging from 0 to 1), with models closer to 1 being better structures. This confidence score corresponds to predicted IDDT score using DeepAccNet [31]. For I-TASSER and C-I-TASSER, default parameters were used in predicting the 3D structure for AgTre. I-TASSER and C-I-TASSER output C-scores within the range between −5 and 2, with a higher C-score signifying higher model confidence.
2.2. Structure Assessment of AgTre Models
Structure assessment of all predicted models in this study (RoseTTAFold, I-Tasser, and C-I-Tasser), as well as available models from SWISS-MODEL and AlphaFold, was carried out using the SWISS-MODEL web server structure assessment tool [32]. Protein model structures from AlphaFold predictions have been integrated into the SWISS-MODEL web server [33,34]. For structural assessment, Qualitative Model Energy Analysis (QMEAN) [35] values around 0 showed reliability, while values ≤−4.0 indicated a low-quality model [36]. A normalised QMEAN4 score compares a predicted model structure with a non-redundant set of PDB structures, providing information on how many standard deviations away from the mean is the predicted model score given a score distribution from a large set of experimentally determined structures. The value varies from |Z-score| < 1 to |Z-score| > 2, with |Z-score| < 1 being closer to the mean, hence a better structure. Ramachandran plot and statistics were obtained, with a model having >98% of its residues in the most favoured regions considered to be of good quality. Similarly, Ramachandran outliers should be <0.2%, Rotamer outliers <1%, number of C-Beta deviations and twisted prolines/non-prolines should be zero. Further assessment was carried out using ERRAT [37] and VERIFY3D [38,39] on SAVES servers (v6.0). For VERIFY3D and ERRAT, models with ≥80% of its amino acid residues having average 3D/1D profile scores ≥ 0.2 and overall quality factor >90%, respectively, were considered to be of good quality.
2.3. Active Site Prediction
The active site residues necessary for the catalytic activity of AgTre were predicted using CASTp [40] and PrankWeb servers [41]. Active site prediction for trehalase from Ae. aegypti (AlphaFold model of Q16V81: AF-Q16V81-F1) was carried out using PrankWeb.
2.4. Ligand Preparation
Validamycin A, trehalozin, trehazolamine, validoxylamine A, casuarine, and uniflorine have been reported to be inhibitors of trehalase in different species. Hence, they were used as the starting compounds to generate more similar structures [9]. However, validamycin A was employed as the control ligand or reference standard due to its commercial availability and reported activity in different insect species. Compounds with similar structures to the control ligand and the previously reported trehalase inhibitors in different species were searched for and retrieved with a 100% search of the PubChem database [42] using a Tanimoto threshold of 80%. This search resulted in a total of 3022 compounds, which were downloaded in their SDF formats. Using the OpenBabel package in PyRx software, the SDF files were minimised using the Universal Force Field (uff) [43] to obtain appropriate proper bond lengths between the different atoms. They were then converted into the pdbqt format and further used for the virtual screening studies.
2.5. Protein Preparation
The best modelled protein structure of AgTre and the downloaded structure of Ae. aegypti (AlphaFold: AF-Q16V81-F1) were prepared using UCSF Chimera [44]. The proteins were minimised using 2000 steepest descent steps and 500 conjugate gradient steps with a conjugate gradient step size of 0.02. AMBER ff14SB charges were added to standard amino acid residues. The prepared PDB file was converted to pdbqt using AutoDock in PyRx.
2.6. Virtual Screening
Virtual screening of the 3022 compounds against AgTre was carried out using AutoDock Vina wizard in PyRx [45]. The vina search space was fitted around the predicted active site residues leading to xyz dimensions of 28.6748, 29.3685, 26.8692, and xyz centre of 0.4168, 7.6228, −20.4605 Å. An exhaustiveness of 8 was used. Similarly, virtual screening of the starting compounds, top hits (obtained from AgTre studies), and optimised compounds (obtained from AgTre studies) against trehalase of Ae. aegypti (AaTre) was carried out. The binding pocket of AaTre was set at xyz dimensions of 25.8307, 27.2200, 30.0916, and xyz centre of −2.1668, −2.0597, and −6.4698 Å. When the virtual screening was completed, binding energies were obtained for the starting compounds, control ligand, and top 9 ligands. Discovery Studio was used to visualise the Trehalase—ligand binding interaction [46].
2.7. Toxicity Screening and Ligand Optimisation
The control ligand, previously reported inhibitors, and top 9 hits were screened for possible ecotoxic effects using admetSAR 2.0 [47], toxicity using ADMETlab 2.0 [48], and their medicinal chemistry properties were determined using SwissADME [49]. ADMETopt [50] was used to optimise the scaffolds of some top hits with the aim of seeing if optimised compounds would have a better binding affinity. A non-toxic constraint (i.e., compounds showing no carcinogenicity, Ames toxicity, acute oral toxicity) was used for optimisation. The new compounds were docked against AgTre and AaTre.
2.8. Molecular Dynamics Simulation
The MD simulation was carried out by using Nanoscale Molecular Dynamics (NAMD) [51] and Visual Molecular Dynamics (VMD) [52]. This helped to determine the stability of the best poses of some of the best hits from the docking studies in the binding site of AgTre. Energy minimisation of 1000 steps was performed to fix the backbone atoms, while the production simulation run was carried out for 1 ns, equivalent to 500,000 steps. The simulation was performed at a constant pressure of 1 atm and temperature of 310 K using Periodic Boundary conditions. The protein structure file (PSF) of the target was generated separately from that of the ligand using VMD, while those of ligands were generated using the Charmm36 forcefield of the Charmm-GUI web server [53]. These were generated to define the bond types, bond angles, atom types, and the number of molecules in the simulation system. The topologies (PDB and PSF) of the protein and ligands were merged, and the complexes were solvated using VMD to generate the cubic water box. The other necessary parameters (time and Periodic Boundary conditions) for the simulation were defined in a script and run using NAMD. The RMSD (Root Mean Square Deviation), RMSF (Root Mean Square Fluctuation), and PCA (Principal Component Analysis) of the simulation results were determined using Bio3D on the Galaxy Europe platform [54], while the hydrogen bond (h-bond) analysis was carried out using VMD software. Graphs were plotted using the plot() function in R-programming software [55].
3. Results
3.1. Structure Prediction and Assessment of AgTre
The 3D structure of AgTre predicted by RoseTTAFold had a confidence score of 0.85, which is close to 1, suggesting a good structure. The best model from I-TASSER had a C-score of −0.22, while that from C-I-TASSER had a C-score of 0.45, suggesting that the model obtained from C-I-TASSER was a better model than that obtained from I-TASSER. I-TASSER and C-I-TASSER models had QMEAN < −4.0, while other models were >−4.0. However, the RoseTTAFold model (−0.10) had QMEAN closest to 0, suggesting it to be of better quality (Table 1). Similarly, it had a normalised QMEAN4 of |Z-score| < 1 as compared to the AlphaFold model with 1 < |Z-score| < 2 (Figure 1A,B). This further suggests that the RoseTTAFold predicted model is the best comparable model with a non-redundant set of PDB structures. The predicted structure from AlphaFold had 96.13% of its residues in Ramachandran favoured, 0.70% Ramachandran outliers, and 0.82% Rotamer outliers compared to 98.06%, 0.35%, and 0%, respectively, observed for the predicted structure from RoseTTAFold in this current study (Table 1). When the RoseTTAFold model and AlphaFold model are aligned, the RMSD, which gives the average deviation between the corresponding atoms between the two proteins, was 0.896 Å when considering 476/570 residues, suggesting the two proteins had similar folds for most parts (Figure 1C). Hence, the structure in this study is comparable to that predicted using AlphaFold. Among all the models tested, the model from RoseTTAFold prediction had the highest number of residues in the Ramachandran favoured region (98.06%). It was the only model with >98% residues in the Ramachandran favoured region (Figure 1D). Based on this parameter, the models from I-TASSER (77.18%) and C-I-TASSER (79.67%) were inferior to all other models. While all models passed ERRAT and VERIFY3D, the RoseTTAFold model had no twisted prolines, cis prolines, rotamer outliers, and C beta deviations as compared to the other models (Table 1). Hence the RoseTTAFold model was observed to be the best model and was used subsequently in this study.

Table 1.
Structural assessment of predicted 3D structures of Anopheles gambiae trehalase (AgTre).
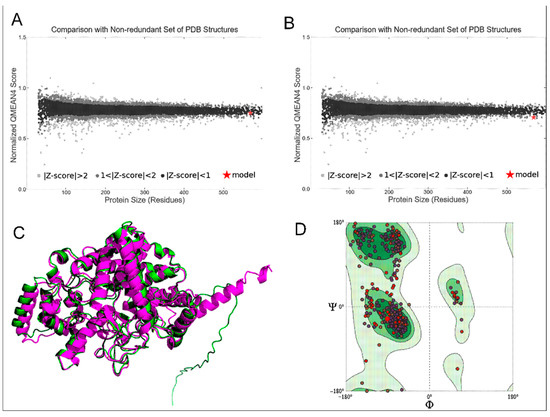
Figure 1.
(A) Normalised QMEAN score of AgTre structure predicted from RoseTTAFold; (B) normalised QMEAN score of AgTre structure predicted by AlphaFold; (C) predicted 3D structure of AgTre. Green: predicted structure from AlphaFold. Purple: predicted structure using RoseTTAFold. RMSD of 0.896 Å considering 476/570 residues; (D) Ramachandran plot of the AgTre structure predicted using RoseTTAFold showing 98.06% of residues in Ramachandran favoured region. AgTre: trehalase of An. gambiae.
3.2. Active Site Prediction
The predicted active site residues of the RoseTTAFold AgTre model and the AaTre model are presented in Table 2. Active site prediction from CASTp yielded a total of 116 pockets, while the prediction from PrankWeb gave 12 pockets for AgTre and 5 pockets for AaTre. The pocket with the highest probability and pocket score was selected for PrankWeb, while the pocket with the largest area and volume was selected for CASTp. The probability for the selected active site pocket for AgTre and AaTre predicted using PrankWeb were 0.948 and 0.855, respectively. For AgTre, 17 amino acids were predicted to be necessary for catalytic activity in the selected pockets from both CASTp and PrankWeb. Four additional residues were predicted to be active site residues by CASTp only. Since the predicted binding pocket from the two programs overlapped, the predicted active site residues from CASTp were used to define the grid box used for molecular docking. Twenty-one (21) residues were predicted as active site residues of AaTre. In the two species, 19 predicted active site residues of trehalase were conserved. This suggests the possibility of both organisms interacting with similar compounds.
Table 2.
Predicted active site residues for trehalase of An. gambiae and Ae. aegypti.
3.3. Molecular Docking
The results obtained from the virtual screening and post-screening studies, ADMET studies, and lead optimisation using ADMETopt are presented in this subsection. Additionally, results comparing binding energies (obtained from virtual screening) between AgTre and AaTre are reported.
3.3.1. Virtual Screening and Post-Screening Studies
The virtual screening studies revealed that all the previously reported trehalase inhibitors that were screened had lower binding energy (i.e., higher affinity) than the control ligand, validamycin A (−5.4 kcal/mol, indicating lowest affinity) for AgTre (Table 3). Among the previously reported trehalase inhibitors, validoxylamine A had the lowest binding energy (−8.8 kcal/mol), indicating the highest affinity for AgTre compared to validamycin A, followed by trehazolin (−7.5 kcal/mol). Uniflorine, casuarine, and trehazolamine had quite similar binding energies for AgTre (−6.9, −6.6, and −6.4 kcal/mol), suggesting that AgTre might have similar affinity for these compounds. Of all the 3022 compounds screened, only 9 compounds had binding energies <−8.8 kcal/mol observed for validoxylamine A (the previously reported trehalase inhibitor with the lowest binding energy). The binding energies were between −8.9 to −9.1 kcal/mol (Table 3). The compounds interacted with different residues in the active site of AgTre through conventional hydrogen bonds, carbon-hydrogen bonds, Pi interactions, and van der Waals interactions. The ligands formed conventional hydrogen bonds, Pi-donor hydrogen bonds, or carbon-hydrogen bonds with two or more of the active site residues Glu258, Asn186, Gln197, Asp292, Ser259, Gly290, Gln438, Arg256, Arg142, Tyr192, Ala287, Phe143, Trp149, Lys26. However, the control ligand, validamycin A, did not bind to any predicted active site residue of the protein. Validamycin A rather formed hydrogen bonds with Gly504, Gln506, Asn307, Ser266 (Table 3, Figures S1 and S2). Other ligands further interacted with other active site residues through van der Waal interactions. Validoxylamine, trehazolamine, 10690241, and 67837201 formed Pi-Sigma interactions with Tyr507, while 25023458 formed Pi-Sigma interactions with Phe143. In addition, validoxylamine, 101104782, 13364642, 25023458, 21021639, and 58618560 had Alkyl/Pi-Alkyl interactions with one or more of Tyr147, Phe143, Arg142, Tyr507, or Lys26. However, the only interaction validamycin A had with an active site residue was a van der Waal interaction with Tyr507. This further indicates that validamycin is a poor binder of AgTre.
Table 3.
Structure, chemical formula, binding energies, and binding interactions of AgTre with the control ligand, previously reported trehalase inhibitors, and the top 9 hits from the virtual screening studies.
3.3.2. ADMET Studies and Lead Optimisation Using ADMETopt
ADME screening of the top nine hit compounds, control ligand, and previously reported inhibitors using SwissADME revealed that they were not blood–brain barrier permeants or CYP 450 inhibitors. The compounds were predicted to be soluble and had no structural alert for Pan-Assay INterference compoundS (PAINS), suggesting that they are not promiscuous binders but would be target-specific [56,57]. Of the top 9 hit compounds, only 10406567, 10690241, and 67837201 had no structural alert for Brenk (Table 4), suggesting that they are non-toxic and metabolically stable [58]. All hit compounds had synthetic accessibility scores lower than validamycin A, which had the highest synthetic accessibility score of 6.34. This suggests that these compounds might be easier to synthesise than validamycin A. The control ligand, starting compounds, and top 9 hits were predicted by admetSAR to be non-toxic to honeybees, crustaceans, and fish. The compounds were predicted to be biodegradable except casuarine and trehazolamine, which had a mild non-biodegradable probability of 0.575 and 0.55, respectively (Table 4). Likewise, employing ADMETlab, all screened compounds were predicted to be non-carcinogenic, cause no irritation or corrosion to the eyes, and cause no skin sensitisation. Predictions by ADMETlab revealed that only 10406567, 10690241, and 67837201 among the top 9 hits had probabilities <0.35 for DILI and respiratory toxicity. Among these three compounds, 10406567 and 10690241 were predicted to have a low probability of being nephrotoxic by admetSAR (probability of 0.4811 and 0.5058, respectively), while 67837201 was not nephrotoxic. Uniflorine (9794258) was predicted to be the safest previously reported compound.
Table 4.
ADMET prediction of control ligand, previously reported trehalase inhibitors, top 9 hits, and optimised compounds.
Further optimisation of the scaffolds of 101104782, 10406567, 10690241, and 67837201 using ADMETopt yielded 120 compounds. These were screened further to select non-toxic compounds based on Brenk alert, a low DILI, and respiratory toxicity probability, yielding 30 compounds. These 30 compounds were docked against AgTre. The best binder was 2-{[6,7-dihydroxy-5-(hydroxymethyl)-3aH,5H,6H,7H,7aH-pyrano[2,3-d][1,3]oxazol-2-yl]amino}-6-(hydroxymethyl)oxane-3,4,5-triol (Compound 1) with a binding energy of −8.9 kcal/mol (Table 5, Figure S3) similar to 67837201, and was predicted not to be nephrotoxic (Table 4). 67837201 and Compound 1 were observed to be the safest compounds with the lowest binding energies for AgTre. Among the optimised compounds, the next top binder after Compound 1 was 4-(hydroxymethyl)-6-{[3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]amino}-hexahydro-[1,3]dioxolo [4,5-c]pyran-2-one (Compound 2) with a binding energy of −8.4 kcal/mol (Table 5), which was predicted to be relatively non-toxic except for a predicted probability of 0.5908 for nephrotoxicity (Table 4).
Table 5.
Structure, chemical formula, binding energies, and binding interactions of AgTre with optimised compounds.
3.3.3. Comparison of Results from AgTre with AaTre
The control compound, previously reported trehalase inhibitors, 101104782, 10406567, 10690241, 67837201, and the 30 optimised compounds were screened against AaTre. The binding energies of these compounds in AaTre and AgTre are presented in Table 6. Similar to the trend observed with AgTre, validoxylamine A had the lowest binding energy (−8.8 kcal/mol) for AaTre, while validamycin A had the highest binding energy (−6.4 kcal/mol). Uniflorine, the safest previously reported trehalase inhibitor, had a binding energy of −7.6 kcal/mol for AaTre and −6.9 kcal/mol for AgTre. The best hit compounds for AaTre were Compound 2 and 10406567 having binding energies of −8.4 and −8.3 kcal/mol, respectively. These compounds had binding energies of −8.4 and 9.0 kcal/mol, respectively, with AgTre. Although Compound 2 and 10406567 were considered safe based on predicted ADMET parameters, they had a slight probability of being nephrotoxic (0.4811 and 0.5908, respectively) (Table 4). The safest compounds (67837201 and Compound 1) with the lowest binding energy for AgTre (−8.9 kcal/mol) had higher binding energy for AaTre (−5.9 and −6.4 kcal/mol, respectively), suggesting they might have a lower affinity for AaTre.
Table 6.
Binding energies of compounds docked against trehalase of An. gambiae and Ae. aegypti.
3.4. Molecular Dynamics Simulation
The control compound (validamycin A), the previously reported trehalase inhibitor with the best affinity for trehalase (validoxylamine A), as well as 67837201, Compound 1, Compound 2, and 10406567 in complex with AgTre, were subjected to MD simulation.
3.4.1. Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF)
RMSD and RMSF results from the molecular dynamics simulation studies are shown in Figure 2A–D, and the individual results are in Figures S4–S8. The stability of protein, indicated by the RMSD changes with respect to Frame No, is presented in Figure 2A. Similarly, the average RMSDs of the protein and standard deviations are shown in Table 7. The binding of validamycin A, 67837201, 10406567, and Compound 1 to AgTre resulted in smaller RMSD changes in the protein compared to the binding of validoxylamine A (the previously reported trehalase inhibitor with the best affinity for trehalase). On the contrary, the binding of Compound 2 resulted in more conformational changes to the protein structure. However, all conformation changes were less than 3 Å, suggesting that the conformation of the protein was stable throughout the MD simulation [59]. RMSF describes the flexibility of each amino acid residue in the protein during the MD simulation. The larger the RMSF value is, the more flexible the residue is. Regarding Figure 2B, the amino acid residues with RMSF > 1.5 Å represent the flexible residues in the loop region of the protein. Similar flexible patterns in the protein were observed upon binding of the different ligands. The RMSDs of the ligands (validamycin A, 67837201, 10406567, Compound 2, and validoxylamine A) are shown in Figure 2C, while that of Compound 1 is shown in Figure 2D. Similarly, the average RMSDs of the ligands and standard deviations are shown in Table 7. The average RMSDs of validoxylamine A, validamycin A, 67837201, and 10406567 reveal that these compounds do not significantly change their orientation during the simulation; hence, they form stable complexes with the protein. This is also confirmed in the histogram (Figure S7). However, Compound 1 was unstable with large changes in its RMSD, having an average RMSD of 5.46 Å with a standard deviation of 1.40 Å (Table 7). Compound 2 showed slight changes early in the simulation but returned to conformations similar to the docking conformation. It had an average RMSD of 1.17 Å and a standard deviation of 0.23 Å. The compound (10406567) exhibited the best stability during the MD simulation with an average RMSD of 0.96 Å and a standard deviation of 0.07 Å. This was followed by Compound 2 and 67837201; 67837201 had an average RMSD of 1.46 Å with a standard deviation of 0.10 Å. The average RMSDs for these compounds was lower than those of validamycin A and validoxylamine A, suggesting that the three compounds maintained relatively stable conformations throughout the simulation period.
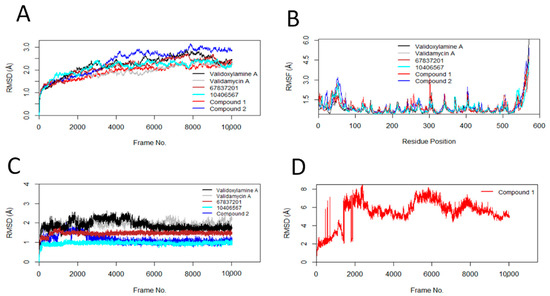
Figure 2.
Conformational changes, residue flexibilities of the protein, and conformational changes of the ligand. (A) Protein Cα-RMSDs during MD simulation with ligand; (B) protein Cα-RMSFs; (C) ligand RMSDs; (D) RMSD of Compound 1.
Table 7.
Average RMSD and standard deviation of bound protein and ligands.
3.4.2. Principal Component Analyses (PCA)
For validoxylamine A, validamycin A, 67837201, 10406567, Compounds 2 and 1, the first three principal components covered 61.3, 54.2, 61, 56.2, 72, and 60.9% of the total variance, respectively, as shown in the eigenvalue rank plots in Figure 3. This suggests that the molecular dynamics simulation procedure captured the major or dominant motions rather than the much less dominant ones. Considering the first three principal components for 67837201, 10406567, Compound 1, and Compound 2, the order of variance was 10406567 < Compound 1 < 67837201 < Compound 2. PC1 accounted for 41.9, 36.8, 40.1, 32.7, 49.7, and 42.4% of the variance for validoxylamine A, validamycin A, 67837201, and 10406567, Compounds 2 and 1. When PC1 alone was considered, the order of variance was 10406567 < 67837201 < Compound 1 < Compound 2. Of the hit compounds screened, Compound 2 had the highest variance, while 10406567 had the lowest variance either when PC1 only or the first three principal components were considered.
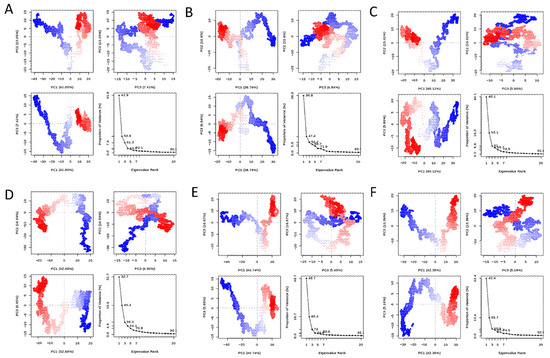
Figure 3.
PCA plots of the protein backbone in the complex, comprising graphs of PC2 vs PC1, PC2 vs PC3, PC3 vs PC1, and an eigenvalue rank plot with the cumulative variance annotated for each data point. (A) Validoxylamine A; (B) validamycin A; (C) 67837201; (D) 10406567; (E) Compound 2; (F) Compound 1.
3.4.3. Hydrogen Bond Analyses
The percentage occupancy of the h-bond >10% between the compounds and amino acid residues of the protein is presented in Table 8. For validoxylamine A, 67837201, and 10406567, Compound 1, and Compound 2, multiple hydrogen bonds were identified between the active site of the protein and the ligand. However, validamycin A did not form hydrogen bonds with any of the active side residues throughout the simulation. The compounds formed h-bonds with similar residues they interacted with in the molecular docking studies (either through hydrogen bond, Pi, unfavourable donor–donor, or van der Waals interactions). Compounds 1 and 2 formed hydrogen bonds with Arg195 (46.26%) and Asp150 (57.72%), respectively, which were not observed in the molecular docking studies. The hydrogen bond interaction with Asp292 was persistent for validoxylamine A (75.46%) and 10406567 (54.56%), while it had an occupancy of 22.62% for 67837201. However, it had an h bond occupancy of <10% for Compound 1 (5.92%) and 2 (6.87%). Glu258 had an h-bond occupancy of 59.33% for 67837201, Asn187 had an occupancy of 58.67% for Compound 1, and Asp150 had an occupancy of 57.72% for Compound 2. Hydrogen bond interactions for all the compounds with other residues were also observed, but these were not persistent (<10%) and were only present for a minority of the time-lapse of the simulation.
Table 8.
Percentage occupancy of the h-bond.
4. Discussion
In this study, validamycin A was observed to have a lower affinity for AgTre and AaTre than validoxylamine A and other previously reported compounds tested (Table 3 and Table 6). These results support previous studies in other organisms suggesting validamycin A to be a poor binder of trehalase compared to validoxylamine A. For example, Asano et al. [60] reported an IC50 of 72 μM for validamycin A in Rizochtonia solani, compared to an IC50 of 140 nM for validoxylamine A. Similarly, validoxylamine A has been observed to have a better inhibitory effect on trehalase than validamycin A in Spodoptera litura (IC50 of 48 nM vs. 370 nM, respectively), porcine intestine (14 nM vs. 120 nM, respectively) [61], porcine kidney (2.4 nM vs. 250 µM) [62], and termites (3.2 μM vs. 402 μM) [63]. In a study by Asano et al. [64], injection of 10 µg of validoxylamine A in the last instar larvae of Spodoptera litura resulted in 70% death in prepupae and 30% abnormal pupae, with no normal pupae or adult being formed. However, injection of validamycin A resulted in 25% death in prepupae, 30% abnormal pupae, and 45% normal pupae, although only 11% of the normal pupae emerged as adults [64]. These studies affirm that validoxylamine A is a better inhibitor of trehalase than validamycin A, corroborating the observation in this study. In this present study, trehazolin (binding energy of −7.5 kcal/mol) was observed to be a better binder of AgTre than validamycin A. Ando et al. [65] reported trehazolin to have an IC50 of 66 nM in Rizochtonia solani compared to IC50 of 72 μM for validamycin A, and 140 nM for validoxylamine A reported earlier by Asano et al. [60], thus suggesting trehazolin is a better inhibitor than validamycin A and validoxylamine A. However, in porcine kidney, Kyosseva et al. [62] reported trehazolin to have an IC50 of 16 nM compared to 2.4 nM for validoxylamine A and 250 µM for validamycin A. This suggests that describing trehazolin as a better trehalase inhibitor when compared to validoxylamine might be organism-dependent. In this current study, validoxylamine was observed to be the best inhibitor of AgTre among the previously reported inhibitors, followed by trehazolin. Uniflorine, trehazolamine, and casuarine had quite similar binding affinities for AgTre, while validamycin A had the least affinity (Table 3). Unlike other compounds which interacted with active site residues of AgTre, validamycin A interacted with other residues (Table 3, Figure S1). Unlike AgTre, in which validamycin A had van der Waal interactions with tyrosine 507 only in the active site, validamycin A had a carbon hydrogen bond with Arg 193, van der Waals interactions with Glu554, Lys77, and Tyr555 in the active site of AaTre. However, conventional hydrogen bonds were formed with the non-active site residues Gly192, Glu313, and Arg312. In both organisms, validamycin A had the highest binding energy among the tested ligands (Table 6). This might explain the low larvicidal activity observed with validamycin treatment in Ae. aegypti by Marten et al. [19]. MD simulation was employed to investigate the stability of the protein–ligand complexes in a flexible system, unlike molecular docking, which assumes that the protein and ligand are in a rigid conformation in most cases [66]. Despite the low affinity for trehalase and not interacting much with active site residues, validamycin A was relatively stable during AgTre-validamycin A MD simulation (Figure 2C, Table 7). It had an h-bond occupancy of 33.97% for Glu262 and 13.03% for Gly504 (Table 8), while the h-bonds formed with other residues persisted below 10% during the whole simulation. These results further corroborate validamycin A as a poor binder of AgTre. This suggests that these other molecules, aside from validamycin A, could serve as better insecticides for malaria vector control.
Uniflorine, the safest previously reported compound showing no predicted toxicity (Table 4), had a binding energy of −6.9 kcal/mol for AgTre and −7.6 kcal/mol for AaTre (Table 6). Uniflorine has been reported to show >5649 selectivity for C. riparius trehalase (177 ± 18 nM) as compared to trehalase from porcine kidney (>1 mM) [67]. Uniflorine is a weaker binder of trehalase from porcine kidney when compared to validoxylamine A, for which an IC50 of 2.4 nM has been reported, but a better binder than validamycin A, for which an IC50 of 250 μM was reported [62]. In this present study, a similar trend was observed with uniflorine having a lower affinity for AgTre and AaTre than validoxylamine A (−8.8 kcal/mol) but a better affinity than validamycin A in both organisms. Considering the high selectivity of uniflorine and its safety, it could be further tested as a possible compound for malaria vector control.
In this study, Compounds 1 and 67837201 were the hit compounds with no toxicity and yet low binding energy for AgTre (Table 3, Table 4 and Table 5). Their binding energies were higher than that of uniflorine, suggesting that they are better binders of AgTre. RMSD results from further examination of the stability of their protein–ligand complexes by MD simulation suggested that Compound 1 might have a binding energy different from that observed in the molecular docking studies as the compound was unstable during the simulation (Figure 2D). This highlights the importance of following up molecular docking studies with MD simulation, as MD simulation has been reported to help separate false positive hits from true positive hits obtained from molecular docking studies [21]. 67837201 was stable during the simulation (Figure 2C); however, unlike in AgTre, where a binding energy of −8.9 kcal/mol was observed during docking studies, it had a higher binding energy for AaTre (−5.9 kcal/mol). Both 10406567 and Compound 2 had good affinity for trehalase of both organisms (Table 6) and displayed stability throughout the simulation (Figure 2C), suggesting they could be excellent compounds for vector control. Further experimental studies to assess their toxicity are necessary, as both were predicted to have a slight probability of being nephrotoxic (Table 4). Similarly, their selectivity for these target organisms needs to be compared to that of non-target organisms in further experimental investigations. However, it has been reported that the disease arising from trehalase inhibition in humans is a mild disease, which can be reversed by avoiding the consumption of mushrooms [68], suggesting trehalase inhibitors as safer alternatives for vector control compared to, for example, organophosphates. Although trehalase enzymes are present in bacteria, fungi, and arthropods [69], uniflorine, 67837201, 10406567, and Compound 2 were predicted to be non-toxic to honeybees, crustaceans, and aquatic life. While these compounds were screened in silico and simulation performed at 310 K, reports of in vivo testing of validamycin A and validoxylamine A in different studies give confidence that the compounds identified in this study would be effective in vivo despite different climatic conditions in which mosquitoes may exist. Experimental validation remains to be carried out as the next future step.
5. Conclusions
This study screened trehalase inhibitors against AgTre to identify inhibitors with better affinity than validamycin A, which could be developed further for malaria vector control. The study proposes uniflorine, 67837201, 10406567, and Compound 2 as possible compounds for malaria vector control. These compounds also bind to AaTre; as such, they can be used to control Aedes aegypti, thus preventing transmission of several other diseases such as Mayaro and dengue fever. The trehalase inhibitors can be applied to breeding sites, as they could affect the development of mosquitoes and prevent their flight to humans, consequently preventing the transmission of mosquito-borne diseases.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/insects13111070/s1, Figure S1: Binding interaction of AgTre with previously reported inhibitors with docked using vina in PyRx, visualised in Discovery Studio, (A) residue glutamate 258, asparagine 186, glutamine 197, and aspartate 292 form conventional hydrogen bonds represented in green dotted lines with validoxylamine A; (B) residue serine 259, glutamate 258, and glutamine 197 form conventional hydrogen bonds represented in green dotted lines with trehazolin; (C) residue glutamate 258, glycine 290, glycine 197, and aspartate 292 form conventional hydrogen bonds represented in green dotted lines with uniflorine; (D) glutamate 258, asparagine 186, glutamine 197, glutamine 438 and aspartate 292 form conventional hydrogen bonds represented in green dotted lines with casuarine; (E) aspartate 292 forms conventional hydrogen bonds represented in green dotted lines with trehazolamine; (F) glycine 504, glutamine 506, asparagine 307, and serine 266 form conventional hydrogen bonds represented in green dotted lines with validamycin A; Figure S2: Binding interaction of AgTre with top 9 hit compounds docked using vina in PyRx, visualised in Discovery Studio, (A) 101104782; (B) 13364642; (C) 56668330; (D) 10406567; (E) 10690241; (F) 58618560; (G) 21021639; (H) 25023458; (I) 67837201; Figure S3: Binding interaction of AgTre with optimised compounds docked using vina in PyRx, visualised in Discovery Studio, (A) Compound 1; (B) Compound 2; Figure S4: Conformational changes and residue flexibilities of the Protein Cα-RMSDs during MD simulation with (A) validoxylamine A; (B) validamycin A; (C) 67837201; (D) 10406567; (E) Compound 2; (F) Compound 1; Figure S5: RMSD histogram of the Protein Cα-RMSDs during MD simulation with (A) validoxylamine A; (B) validamycin A; (C) 67837201; (D) 10406567; (E) Compound 2; (F) Compound 1; Figure S6: Ligand RMSD during MD simulation (A) validoxylamine A; (B) validamycin A; (C) 67837201; (D) 10406567; (E) Compound 2; (F) Compound 1; Figure S7: Ligand RMSD histogram during MD simulation (A) validoxylamine A; (B) validamycin A; (C) 67837201; (D) 10406567; (E) Compound 2; (F) Compound 1; Figure S8: Protein Cα-RMSFs during MD simulation with (A) validoxylamine A; (B) validamycin A; (C) 67837201; (D) 10406567; (E) Compound 2; (F) Compound 1.
Author Contributions
Conceptualisation: E.O.A.; methodology and analysis, E.O.A. and G.O.O.; writing—original draft preparation: E.O.A.; writing–review and editing: E.O.A., G.O.O., O.O.O., S.F., R.K., and E.A.; supervision, O.O.O., S.F., R.K., and E.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Deutsche Forschungsgemeinschaft (DFG), grant number KO 3678/5-1 and Fogarty National Institutes of Health (NIH) Common Fund, grant number U2RTW010679. SF was supported by Wellcome Trust grant number 220740/Z/20/Z.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Materials.
Acknowledgments
We appreciate Covenant University for the support provided; R.K. and E.A (funding acquisition).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Cribellier, A.; Straw, A.D.; Spitzen, J.; Pieters, R.P.; van Leeuwen, J.L.; Muijres, F.T. Diurnal and nocturnal mosquitoes escape looming threats using distinct flight strategies. Curr. Biol. 2022, 32, 1232–1246.e5. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.L.; Courtenay, O.; Kelly-Hope, L.A.; Scott, T.W.; Takken, W.; Torr, S.J.; Lindsay, S.W. The importance of vector control for the control and elimination of vector-borne diseases. PLoS Negl. Trop. Dis. 2020, 14, e0007831. [Google Scholar] [CrossRef] [PubMed]
- Kaindoa, E.W.; Mmbando, A.S.; Shirima, R.; Hape, E.E.; Okumu, F.O. Insecticide-treated eave ribbons for malaria vector control in low-income communities. Malar. J. 2021, 20, 415. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Weiss, D.; Cameron, E.; Bisanzio, D.; Mappin, B.; Dalrymple, U.; Battle, K.; Moyes, C.; Henry, A.; Eckhoff, P. The effect of malaria control on Plasmodium falciparum in Africa between 2000 and 2015. Nature 2015, 526, 207–211. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Malaria Report 2021; WHO: Geneva, Switzerland, 2021.
- Ogunah, J.A.; Lalah, J.O.; Schramm, K.-W. Malaria vector control strategies. What is appropriate towards sustainable global eradication? Sustain. Chem. Pharm. 2020, 18, 100339. [Google Scholar] [CrossRef]
- Gowelo, S.; McCann, R.S.; Koenraadt, C.J.; Takken, W.; van den Berg, H.; Manda-Taylor, L. Community factors affecting participation in larval source management for malaria control in Chikwawa District, Southern Malawi. Malar. J. 2020, 19, 195. [Google Scholar] [CrossRef]
- Fillinger, U.; Lindsay, S.W. Larval source management for malaria control in Africa: Myths and reality. Malar. J. 2011, 10, 353. [Google Scholar] [CrossRef]
- Matassini, C.; Parmeggiani, C.; Cardona, F. New frontiers on human safe insecticides and fungicides: An opinion on trehalase inhibitors. Molecules 2020, 25, 3013. [Google Scholar] [CrossRef]
- Alexandrino, A.V.; Goto, L.S.; Novo-Mansur, M.T.M. treA codifies for a trehalase with involvement in Xanthomonas citri subsp. citri pathogenicity. PLoS ONE 2016, 11, e0162886. [Google Scholar] [CrossRef]
- Becker, A.; Schlöder, P.; Steele, J.; Wegener, G. The regulation of trehalose metabolism in insects. Experientia 1996, 52, 433–439. [Google Scholar] [CrossRef]
- Paul, M.J.; Primavesi, L.F.; Jhurreea, D.; Zhang, Y. Trehalose metabolism and signaling. Annu. Rev. Plant Biol. 2008, 59, 417. [Google Scholar] [CrossRef] [PubMed]
- D’Adamio, G.; Forcella, M.; Fusi, P.; Parenti, P.; Matassini, C.; Ferhati, X.; Vanni, C.; Cardona, F. Probing the influence of linker length and flexibility in the design and synthesis of new trehalase inhibitors. Molecules 2018, 23, 436. [Google Scholar] [CrossRef] [PubMed]
- Tevatiya, S.; Kumari, S.; Sharma, P.; Rani, J.; Chauhan, C.; Das De, T.; Pandey, K.C.; Pande, V.; Dixit, R. Molecular and functional characterization of Trehalase in the mosquito Anopheles stephensi. Front. Physiol. 2020, 11, 1455. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Dong, Y.; Huang, Y.; Rasgon, J.L.; Agre, P. Impact of trehalose transporter knockdown on Anopheles gambiae stress adaptation and susceptibility to Plasmodium falciparum infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17504–17509. [Google Scholar] [CrossRef] [PubMed]
- García, M.D.; Argüelles, J.C. Trehalase inhibition by validamycin A may be a promising target to design new fungicides and insecticides. Pest Manag. Sci. 2021, 77, 3832–3835. [Google Scholar] [CrossRef] [PubMed]
- Adhav, A.S.; Kokane, S.R.; Joshi, R.S. Functional characterization of Helicoverpa armigera trehalase and investigation of physiological effects caused due to its inhibition by Validamycin A formulation. Int. J. Biol. Macromol. 2018, 112, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Z.; Huang, Y.L.; Lu, Z.J.; Zhang, Q.; Su, H.N.; Du, Y.M.; Yi, L.; Zhong, B.L.; Chen, C.X. Inhibition of trehalase affects the trehalose and chitin metabolism pathways in Diaphorina citri (Hemiptera: Psyllidae). J. Insect Sci. 2021, 28, 718–734. [Google Scholar] [CrossRef]
- Marten, A.D.; Stothard, A.I.; Kalera, K.; Swarts, B.M.; Conway, M.J. Validamycin A delays development and prevents flight in Aedes aegypti (Diptera: Culicidae). J. Med. Entomol. 2020, 57, 1096–1103. [Google Scholar] [CrossRef]
- Logan, C.I. Trehalase Inhibition in Aedes aegypti. Master’s Thesis, Simon Fraser University, Burnaby, BC, Canada, 2008. [Google Scholar]
- Sohraby, F.; Bagheri, M.; Aryapour, H. Performing an in silico repurposing of existing drugs by combining virtual screening and molecular dynamics simulation. In Computational Methods for Drug Repurposing; Humana Press: New York, NY, USA, 2019; pp. 23–43. [Google Scholar]
- Salmaso, V.; Moro, S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef]
- Salmaso, V.; Sturlese, M.; Cuzzolin, A.; Moro, S. DockBench as docking selector tool: The lesson learned from D3R Grand Challenge 2015. J. Comput. Aided Mol. Des. 2016, 30, 773–789. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, X.; Katsuma, S.; Xu, Y.; Shi, L.; Shimada, T.; Wang, H. Duplication and diversification of trehalase confers evolutionary advantages on lepidopteran insects. Mol. Ecol. 2019, 28, 5282–5298. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; De Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Hiranuma, N.; Park, H.; Baek, M.; Anishchenko, I.; Dauparas, J.; Baker, D. Improved protein structure refinement guided by deep learning based accuracy estimation. Nat. Commun. 2021, 12, 1340. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Singh, V.; Patel, D. Structural bioinformatics. In Essentials of Bioinformatics, Volume I; Springer: Cham, Denmark, 2019; pp. 169–199. [Google Scholar]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. [20] VERIFY3D: Assessment of protein models with three-dimensional profiles. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1997; Volume 277, pp. 396–404. [Google Scholar]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.; Goddard III, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- BIOVIA Dassault Systèmes. Discovery Studio, 2022; Dassault Systèmes: San Diego, CA, USA, 2022. [Google Scholar]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Sun, L.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. ADMETopt: A web server for ADMET optimization in drug design via scaffold hopping. J. Chem. Inf. Model. 2018, 58, 2051–2056. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2021. [Google Scholar]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem Chem. Enabling Drug Discov. 2008, 3, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Zou, R.; Chan, H.S.; Li, H.; Yuan, S. MolADI: A web server for automatic analysis of protein–small molecule dynamic interactions. Molecules 2021, 26, 4625. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Yamaguchi, T.; Kameda, Y.; Matsui, K. Effect of validamycins on glycohydrolases of Rhizoctonia solani. J. Antibiot. 1987, 40, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Kameda, Y.; Asano, N.; Yamaguchi, T.; Matsui, K. Validoxylamines as trehalase inhibitors. J. Antibiot. 1987, 40, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Kyosseva, S.V.; Kyossev, Z.N.; Elbein, A.D. Inhibitors of pig kidney trehalase. Arch. Biochem. Biophys. 1995, 316, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.-Q.; Zheng, Y.-G. Inhibitory effects of validamycin compounds on the termites trehalase. Pestic. Biochem. Physiol. 2009, 95, 28–32. [Google Scholar] [CrossRef]
- Asano, N.; Takeuchi, M.; Kameda, Y.; Matsui, K.; Kono, Y. Trehalase inhibitors, validoxylamine A and related compounds as insecticides. J. Antibiot. 1990, 43, 722–726. [Google Scholar] [CrossRef]
- Ando, O.; Kifune, M.; Nakajima, M. Effects of trehazolin, a potent trehalase inhibitor, on Bombyx mori and plant pathogenic fungi. Biosci. Biotechnol. Biochem. 1995, 59, 711–712. [Google Scholar] [CrossRef]
- Li, F.; Venthur, H.; Wang, S.; Homem, R.A.; Zhou, J.-J. Evidence for the involvement of the chemosensory protein Agoscsp5 in resistance to insecticides in the cotton aphid, Aphis gossypii. Insects 2021, 12, 335. [Google Scholar] [CrossRef]
- D’Adamio, G.; Sgambato, A.; Forcella, M.; Caccia, S.; Parmeggiani, C.; Casartelli, M.; Parenti, P.; Bini, D.; Cipolla, L.; Fusi, P. New synthesis and biological evaluation of uniflorine A derivatives: Towards specific insect trehalase inhibitors. Org. Biomol. Chem. 2015, 13, 886–892. [Google Scholar] [CrossRef]
- Richards, A.; Krakowka, S.; Dexter, L.; Schmid, H.; Wolterbeek, A.; Waalkens-Berendsen, D.; Shigoyuki, A.; Kurimoto, M. Trehalose: A review of properties, history of use and human tolerance, and results of multiple safety studies. Food Chem. Toxicol. 2002, 40, 871–898. [Google Scholar] [CrossRef]
- Argüelles, J.-C. Why can’t vertebrates synthesize trehalose? J. Mol. Evol. 2014, 79, 111–116. [Google Scholar] [CrossRef] [PubMed]




















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).