
Molecular Characterization of a Clade 2.3.4.4b H5N1 High Pathogenicity Avian Influenza Virus from a 2022 Outbreak in Layer Chickens in the Philippines
 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Quantitative RT-PCR
2.2. Whole-Genome Sequencing
2.3. Sequence Assembly
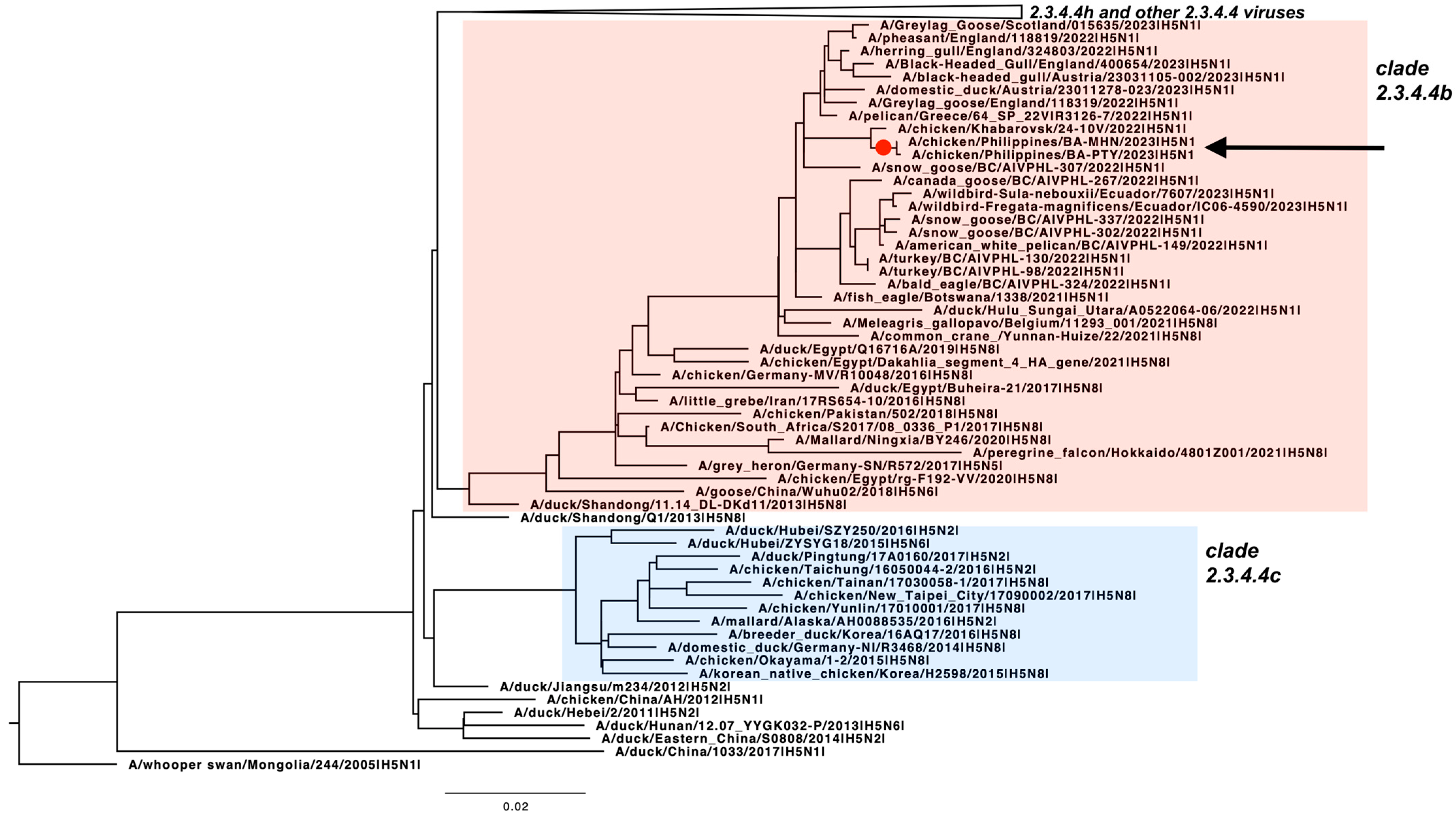
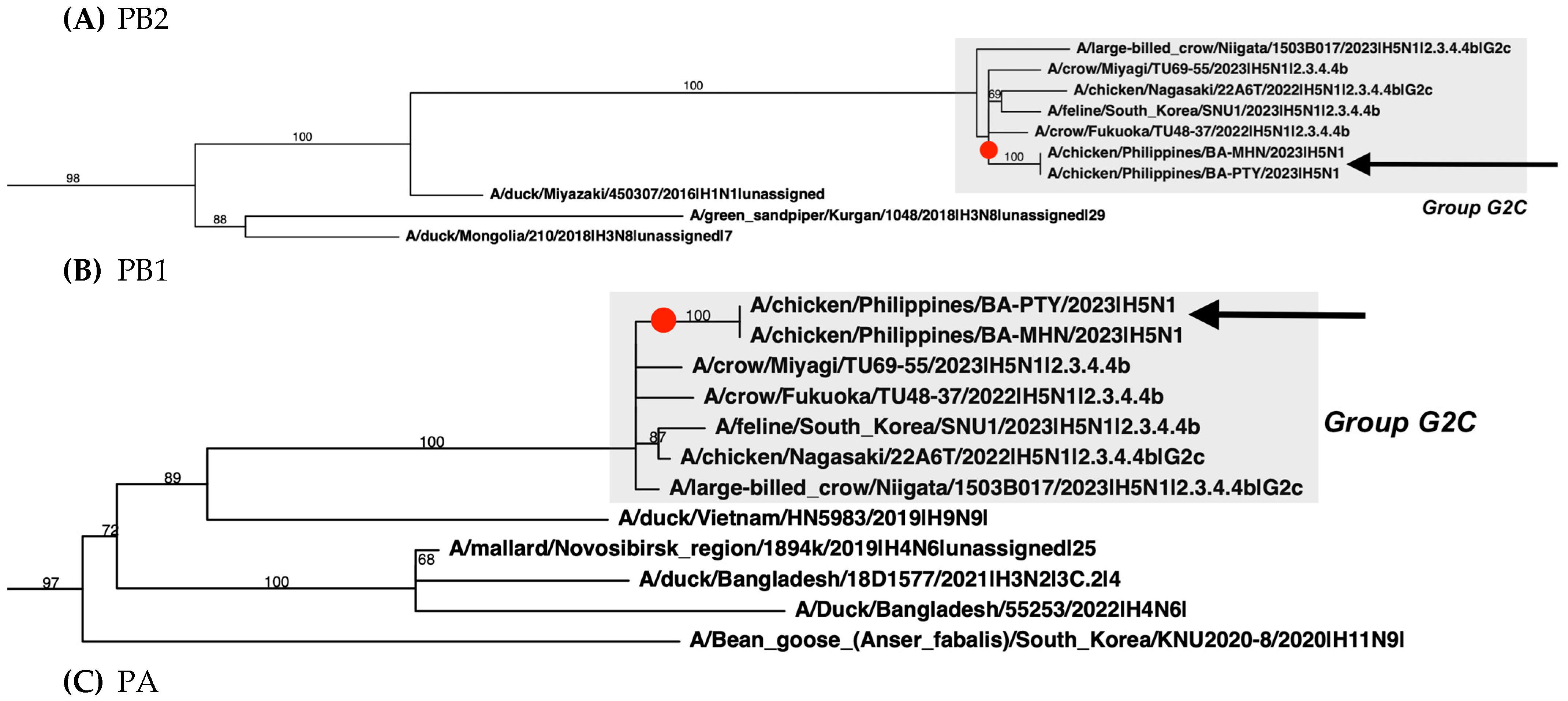
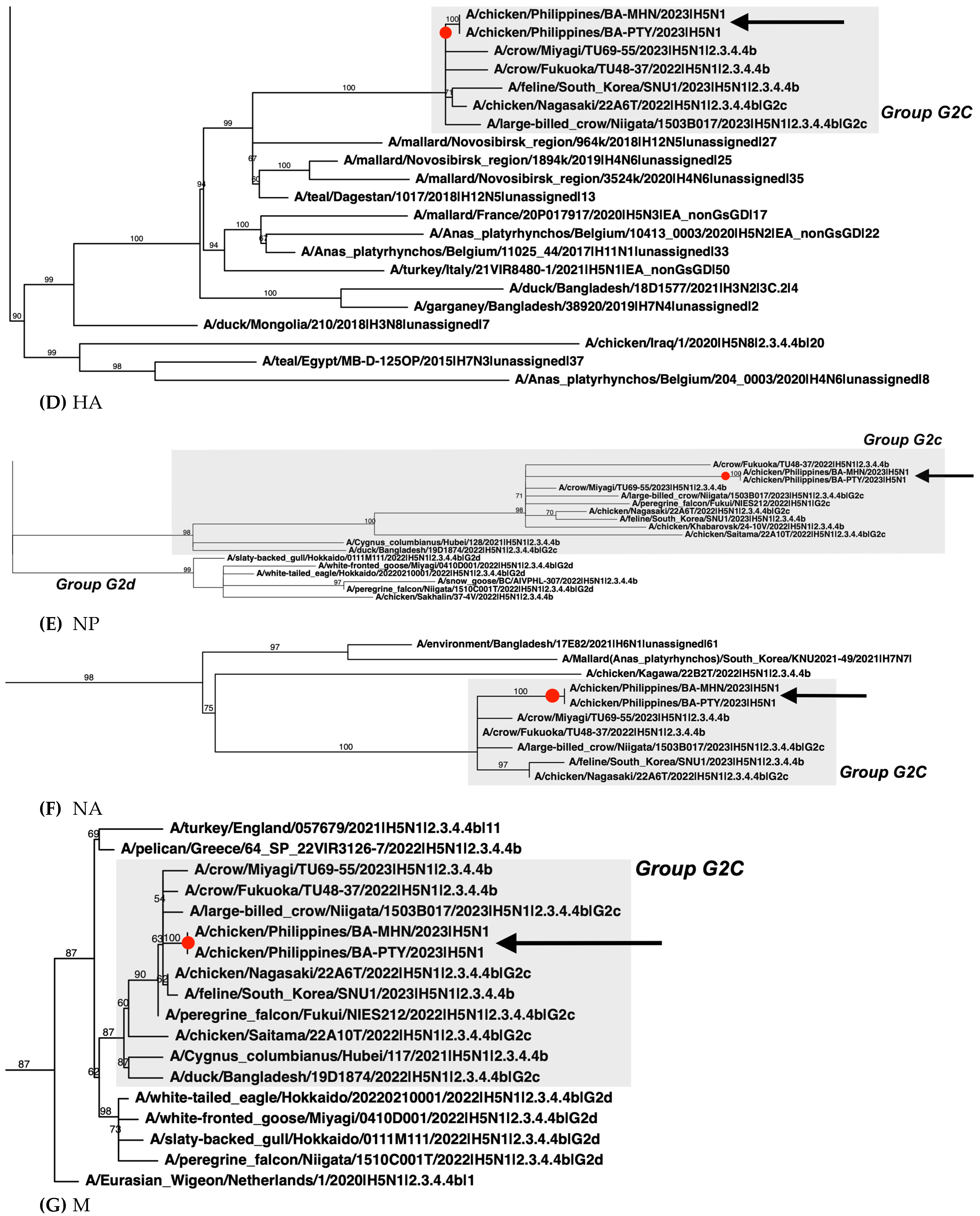
2.4. Phylogenetic Analysis
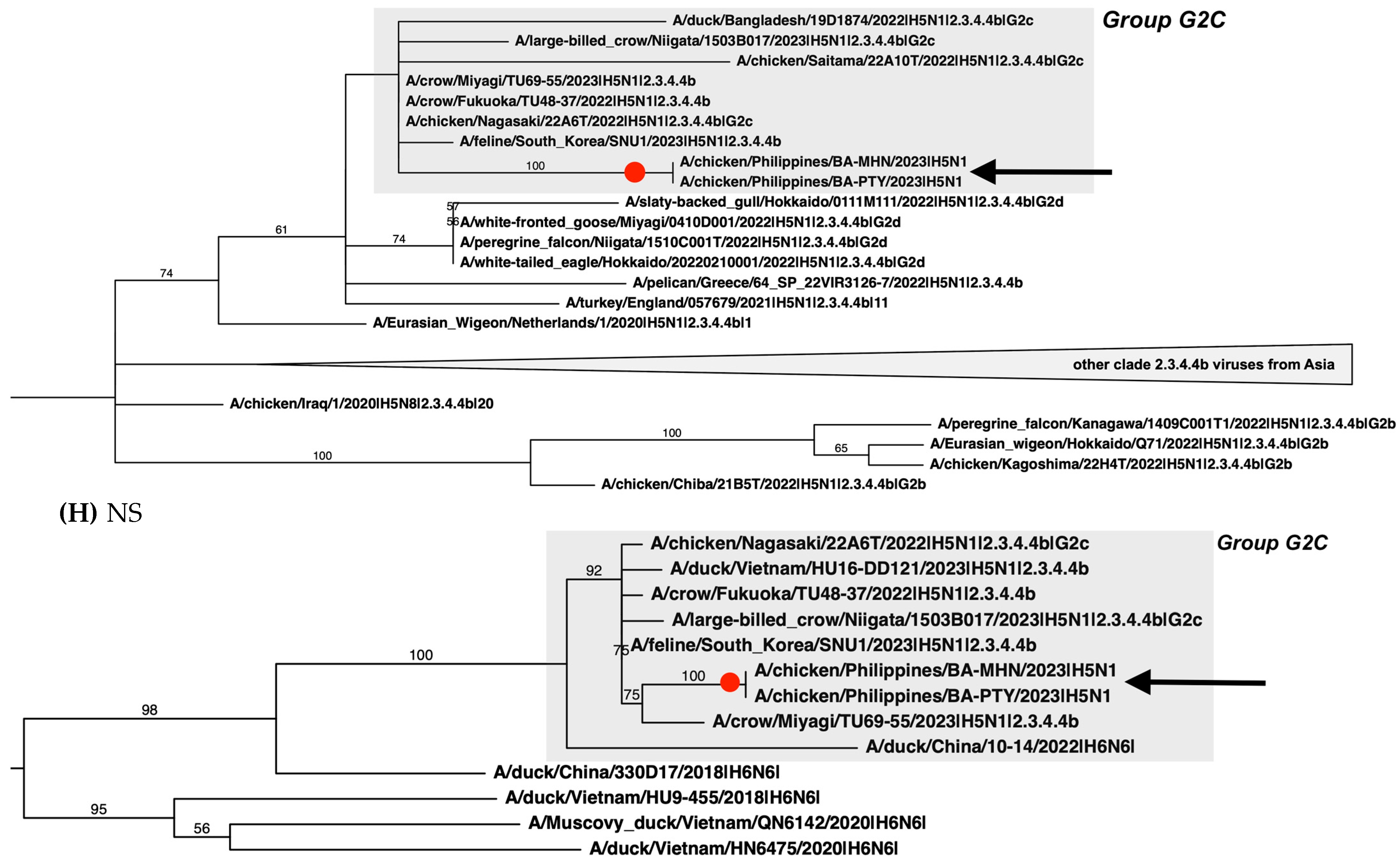
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Long, J.S.; Mistry, B.; Haslam, S.M.; Barclay, W.S. Host and viral determinants of influenza A virus species specificity. Nat. Rev. Microbiol. 2019, 17, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Gao, R.; Lv, Q.; Huang, S.; Zhou, Z.; Yang, L.; Li, X.; Zhao, X.; Zou, X.; Tong, W.; et al. Human infection with a novel, highly pathogenic avian influenza A (H5N6) virus: Virological and clinical findings. J. Infect. 2016, 72, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Bertran, K.; Kwon, J.H.; Swayne, D.E. Evolution, global spread, and pathogenicity of highly pathogenic avian influenza H5Nx clade 2.3.4.4. J. Vet. Sci. 2017, 18, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.H.; Bertran, K.; Lee, D.H.; Criado, M.F.; Killmaster, L.; Pantin-Jackwood, M.J.; Swayne, D.E. Diverse infectivity, transmissibility, and pathobiology of clade 2.3.4.4 H5Nx highly pathogenic avian influenza viruses in chickens. Emerg. Microbes Infect. 2023, 12, 2218945. [Google Scholar] [CrossRef] [PubMed]
- Charostad, J.; Rezaei Zadeh Rukerd, M.; Mahmoudvand, S.; Bashash, D.; Hashemi, S.M.A.; Nakhaie, M.; Zandi, K. A comprehensive review of highly pathogenic avian influenza (HPAI) H5N1: An imminent threat at doorstep. Travel. Med. Infect. Dis. 2023, 55, 102638. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. f. A. WAHIS: World Animal Health Information System. Available online: https://wahis.woah.org/ (accessed on 25 August 2023).
- Thielen, P. Influenza Whole Genome Sequencing with Integrated Indexing on Oxford Nanopore Platforms V.1. Available online: https://slack.protocols.io:8443/view/influenza-whole-genome-sequencing-with-integrated-wykffuw.html (accessed on 15 July 2023).
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Takadate, Y.; Mine, J.; Tsunekuni, R.; Sakuma, S.; Kumagai, A.; Nishiura, H.; Miyazawa, K.; Uchida, Y. Genetic diversity of H5N1 and H5N2 high pathogenicity avian influenza viruses isolated from poultry in Japan during the winter of 2022–2023. Virus Res. 2024, 347, 199425. [Google Scholar] [CrossRef]
- Fusaro, A.; Zecchin, B.; Giussani, E.; Palumbo, E.; Agüero-García, M.; Bachofen, C.; Bálint, Á.; Banihashem, F.; Banyard, A.C.; Beerens, N.; et al. High pathogenic avian influenza A(H5) viruses of clade 2.3.4.4b in Europe-Why trends of virus evolution are more difficult to predict. Virus Evol. 2024, 10, veae027. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed]
- Nabeshima, K.; Takadate, Y.; Soda, K.; Hiono, T.; Isoda, N.; Sakoda, Y.; Mine, J.; Miyazawa, K.; Onuma, M.; Uchida, Y. Detection of H5N1 High Pathogenicity Avian Influenza Viruses in Four Raptors and Two Geese in Japan in the Fall of 2022. Viruses 2023, 15, 1865. [Google Scholar] [CrossRef] [PubMed]
- Mine, J.; Takadate, Y.; Kumagai, A.; Sakuma, S.; Tsunekuni, R.; Miyazawa, K.; Uchida, Y. Genetics of H5N1 and H5N8 High-Pathogenicity Avian Influenza Viruses Isolated in Japan in Winter 2021–2022. Viruses 2024, 16, 358. [Google Scholar] [CrossRef] [PubMed]
- Luczo, J.M.; Tachedjian, M.; Harper, J.A.; Payne, J.S.; Butler, J.M.; Sapats, S.I.; Lowther, S.L.; Michalski, W.P.; Stambas, J.; Bingham, J. Evolution of high pathogenicity of H5 avian influenza virus: Haemagglutinin cleavage site selection of reverse-genetics mutants during passage in chickens. Sci. Rep. 2018, 8, 11518. [Google Scholar] [CrossRef] [PubMed]
- Hatta, M.; Hatta, Y.; Kim, J.H.; Watanabe, S.; Shinya, K.; Nguyen, T.; Lien, P.S.; Le, Q.M.; Kawaoka, Y. Growth of H5N1 Influenza A Viruses in the Upper Respiratory Tracts of Mice. PLoS Pathog. 2007, 3, e133. [Google Scholar] [CrossRef]
- Bogs, J.; Kalthoff, D.; Veits, J.; Pavlova, S.; Schwemmle, M.; Mänz, B.; Mettenleiter, T.C.; Stech, J. Reversion of PB2-627E to -627K during replication of an H5N1 Clade 2.2 virus in mammalian hosts depends on the origin of the nucleoprotein. J. Virol. 2011, 85, 10691–10698. [Google Scholar] [CrossRef]
- Tarendeau, F.; Crepin, T.; Guilligay, D.; Ruigrok, R.W.H.; Cusack, S.; Hart, D.J. Host Determinant Residue Lysine 627 Lies on the Surface of a Discrete, Folded Domain of Influenza Virus Polymerase PB2 Subunit. PLoS Pathog. 2008, 4, e1000136. [Google Scholar] [CrossRef]
- Fan, S.; Hatta, M.; Kim, J.H.; Halfmann, P.; Imai, M.; Macken, C.A.; Le, M.Q.; Nguyen, T.; Neumann, G.; Kawaoka, Y. Novel residues in avian influenza virus PB2 protein affect virulence in mammalian hosts. Nat. Commun. 2014, 5, 5021. [Google Scholar] [CrossRef]
- Gabriel, G.; Dauber, B.; Wolff, T.; Planz, O.; Klenk, H.D.; Stech, J. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc. Natl. Acad. Sci. USA 2005, 102, 18590–18595. [Google Scholar] [CrossRef]
- Tada, T.; Suzuki, K.; Sakurai, Y.; Kubo, M.; Okada, H.; Itoh, T.; Tsukamoto, K. NP body domain and PB2 contribute to increased virulence of H5N1 highly pathogenic avian influenza viruses in chickens. J. Virol. 2011, 85, 1834–1846. [Google Scholar] [CrossRef]
- Salvador, R.; Tanquilut, N.; Na Lampang, K.; Chaisowwong, W.; Pfeiffer, D.; Punyapornwithaya, V. Identification of High-Risk Areas for the Spread of Highly Pathogenic Avian Influenza in Central Luzon, Philippines. Vet. Sci. 2020, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Suttie, A.; Deng, Y.M.; Greenhill, A.R.; Dussart, P.; Horwood, P.F.; Karlsson, E.A. Inventory of molecular markers affecting biological characteristics of avian influenza A viruses. Virus Genes 2019, 55, 739–768. [Google Scholar] [CrossRef] [PubMed]
- Wasilenko, J.L.; Sarmento, L.; Pantin-Jackwood, M.J. A single substitution in amino acid 184 of the NP protein alters the replication and pathogenicity of H5N1 avian influenza viruses in chickens. Arch. Virol. 2009, 154, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Yuan, S.; Zhang, K.; Singh, K.; Ma, Q.; Zhou, J.; Chu, H.; Zheng, B.-J. PB2 substitutions V598T/I increase the virulence of H7N9 influenza A virus in mammals. Virology 2017, 501, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Cui, P.; Song, Y.; Qi, Y.; Li, X.; Qi, W.; Xu, C.; Jiao, P.; Liao, M. PB2 segment promotes high-pathogenicity of H5N1 avian influenza viruses in mice. Front. Microbiol. 2015, 6, 73. [Google Scholar] [CrossRef]
- Su, Y.; Yang, H.-Y.; Zhang, B.-J.; Jia, H.-L.; Tien, P. Analysis of a point mutation in H5N1 avian influenza virus hemagglutinin in relation to virus entry into live mammalian cells. Arch. Virol. 2008, 153, 2253–2261. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Segment | Size (bp) | GC Content (%) | Strain Name for Top BLAST Hit | Identity at Nucleotide Level (%) | GenBank Accession No. |
|---|---|---|---|---|---|
| PB2 | 2323 | 43.7 | A/crow/Miyagi/TU69-55/2023(H5N1) | 99.8 | LC765306 |
| PB1 | 2323 | 43.7 | A/crow/Miyagi/TU69-55/2023(H5N1) | 99.4 | LC765307 |
| PA | 2214 | 44.9 | A/crow/Miyagi/TU69-55/2023(H5N1) | 99.6 | LC765308 |
| HA | 1757 | 41.9 | A/crow/Miyagi/TU69-55/2023(H5N1) | 99.5 | LC765309 |
| NP | 1544 | 47.3 | A/crow/Fukuoka/TU48-37/2022(H5N1) | 99.6 | LC765294 |
| NA | 1436 | 44.1 | A/crow/Fukuoka/TU48-37/2022(H5N1) | 99.4 | LC765295 |
| M | 1002 | 49.5 | A/crow/Fukuoka/TU48-37/2022(H5N1) | 99.9 | LC765296 |
| NS | 864 | 43.1 | A/feline/South Korea/SNU1/2023(H5N1) | 99.3 | OR388766 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baybay, Z.; Montecillo, A.; Pantua, A.; Mananggit, M.; Romo, G.R., Jr.; San Pedro, E.; Pantua, H.; Leyson, C.L. Molecular Characterization of a Clade 2.3.4.4b H5N1 High Pathogenicity Avian Influenza Virus from a 2022 Outbreak in Layer Chickens in the Philippines. Pathogens 2024, 13, 844. https://doi.org/10.3390/pathogens13100844
Baybay Z, Montecillo A, Pantua A, Mananggit M, Romo GR Jr., San Pedro E, Pantua H, Leyson CL. Molecular Characterization of a Clade 2.3.4.4b H5N1 High Pathogenicity Avian Influenza Virus from a 2022 Outbreak in Layer Chickens in the Philippines. Pathogens. 2024; 13(10):844. https://doi.org/10.3390/pathogens13100844
Chicago/Turabian StyleBaybay, Zyne, Andrew Montecillo, Airish Pantua, Milagros Mananggit, Generoso Rene Romo, Jr., Esmeraldo San Pedro, Homer Pantua, and Christina Lora Leyson. 2024. "Molecular Characterization of a Clade 2.3.4.4b H5N1 High Pathogenicity Avian Influenza Virus from a 2022 Outbreak in Layer Chickens in the Philippines" Pathogens 13, no. 10: 844. https://doi.org/10.3390/pathogens13100844
APA StyleBaybay, Z., Montecillo, A., Pantua, A., Mananggit, M., Romo, G. R., Jr., San Pedro, E., Pantua, H., & Leyson, C. L. (2024). Molecular Characterization of a Clade 2.3.4.4b H5N1 High Pathogenicity Avian Influenza Virus from a 2022 Outbreak in Layer Chickens in the Philippines. Pathogens, 13(10), 844. https://doi.org/10.3390/pathogens13100844