
A Polyphasic Approach Reveals Novel Genotypes and Updates the Genetic Structure of the Banana Fusarium Wilt Pathogen

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Isolates
2.2. Cultural, Morphological and Molecular Identification
2.3. Pathogenicity Testing
2.4. Nit-Mutant Generation and VCG Testing
2.5. Multi-Gene Phylogeny
2.6. Genome-Wide Genotyping
2.6.1. DArT Sequencing
2.6.2. Phylogenetic Analysis Based on DArT-Seq Data
2.6.3. Genetic Structure Analysis
2.7. Presence of SIX Gene Homologues in Foc
3. Results
3.1. Cultural and Morphological Identification
3.2. Pathogenicity Testing
3.3. VCG Testing
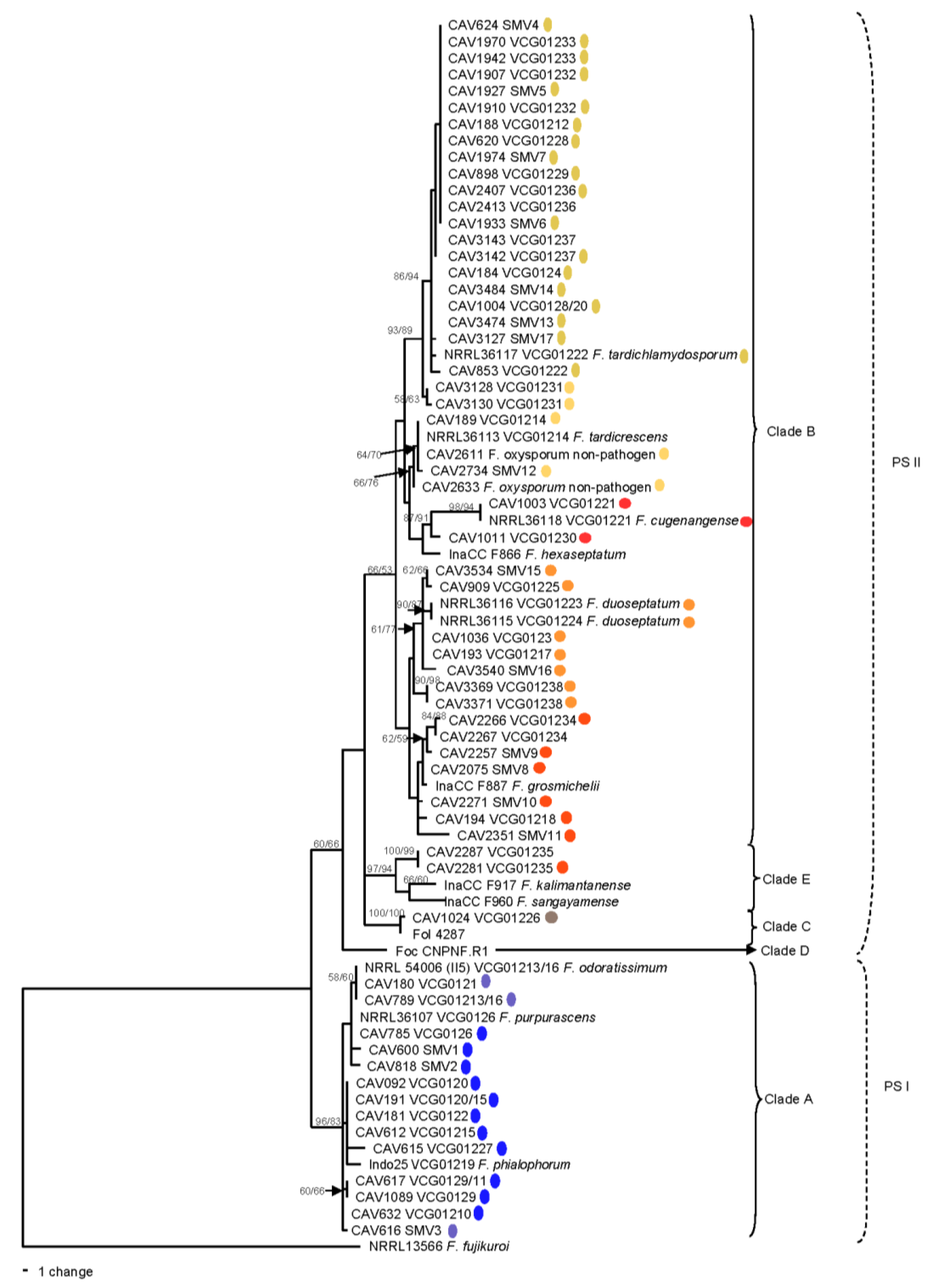
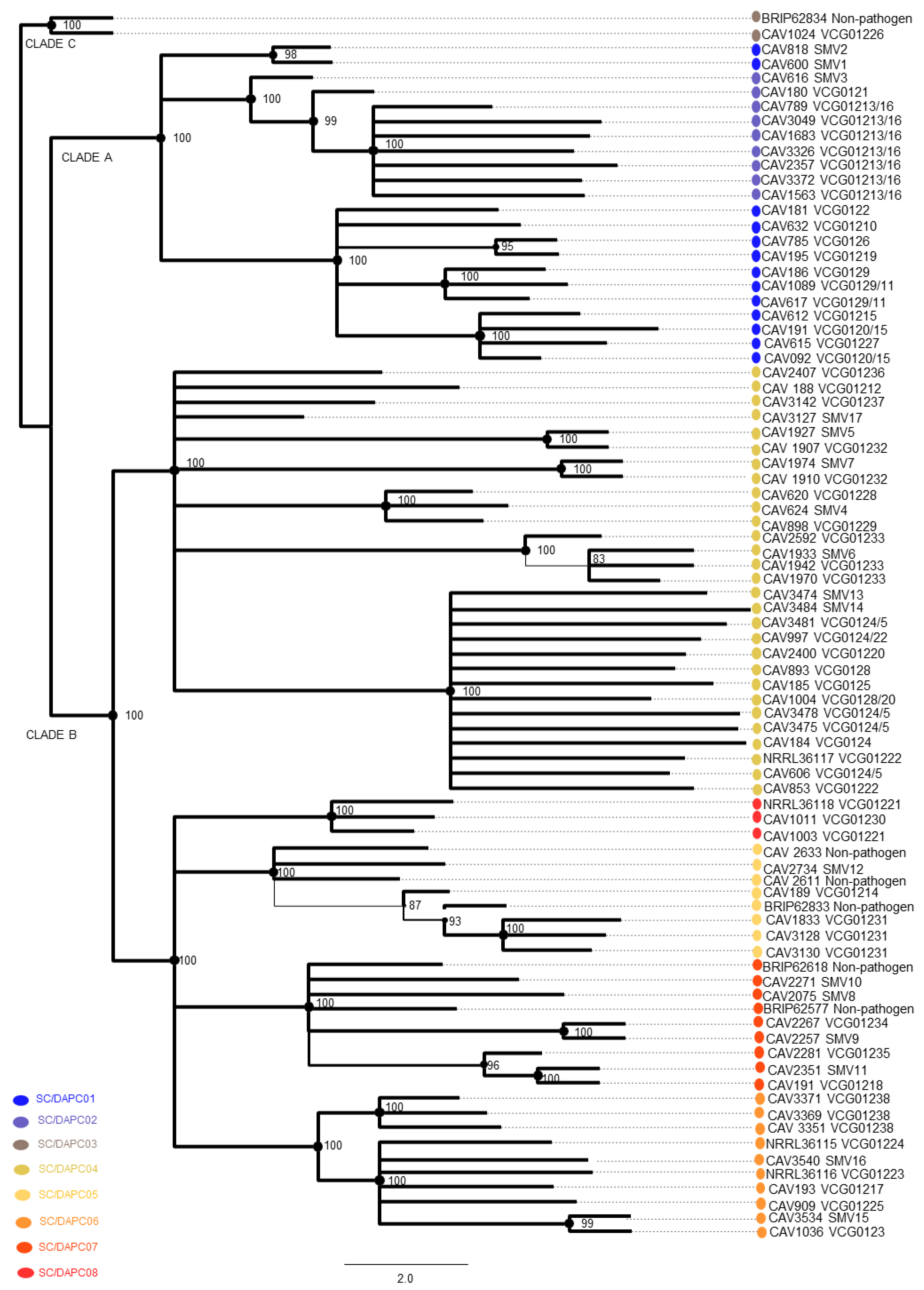
3.4. Multi-Gene Phylogenetic Analysis
3.5. DArT Sequencing
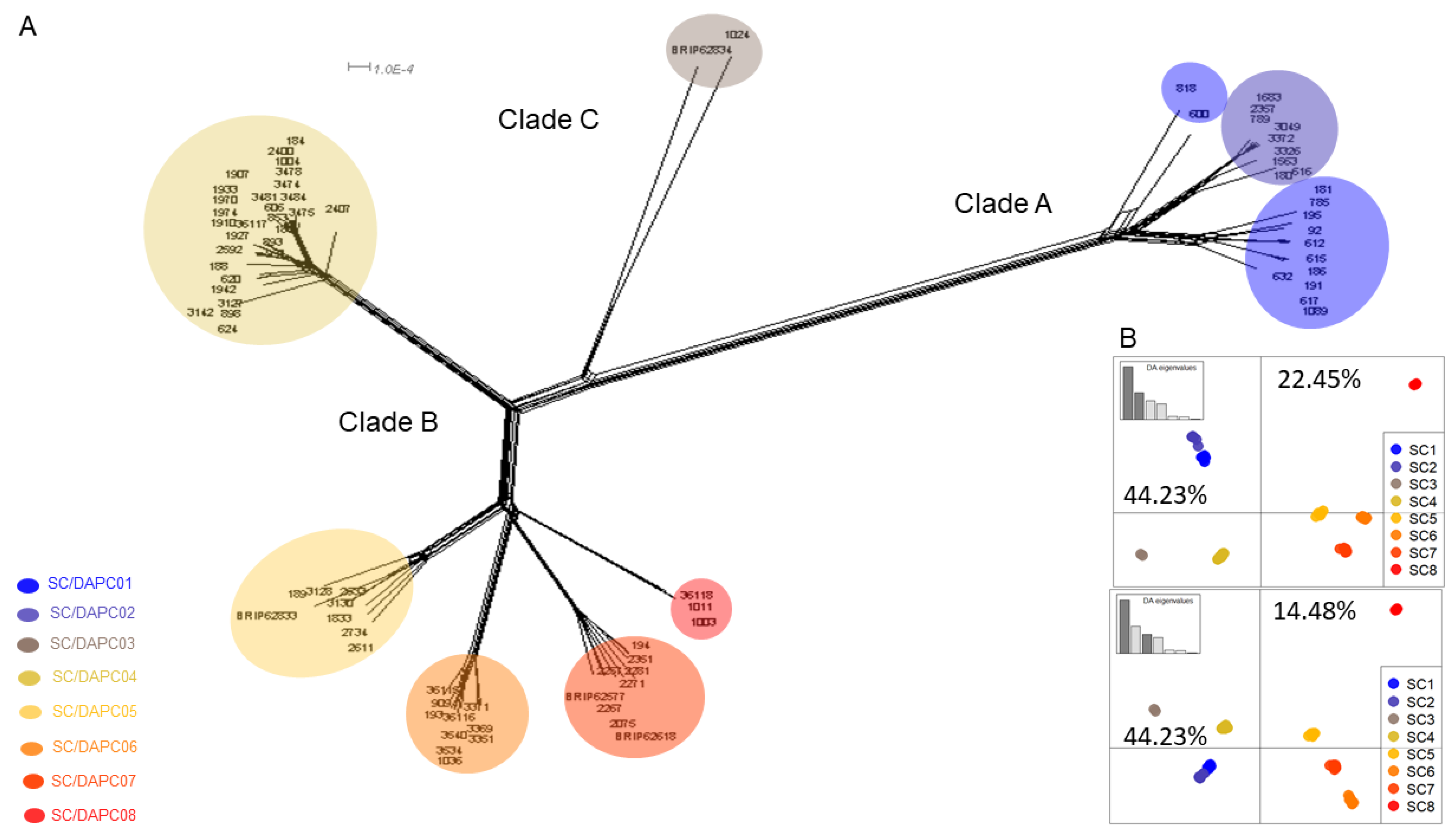
3.6. Genome-Wide Genotyping
3.7. Phylogenetic Analysis of SNP Data
3.8. Correspondence between Multi-Gene and DArT-Seq SNP Phylogenies
3.9. Presence of SIX Gene Homologues in Foc
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leslie, J.F.; Summerell, B.A. The Fusarium Laboratory Manual; Blackwell Publishing: Ames, IA, USA, 2006. [Google Scholar]
- Su, H.J. Fusarial Wilt of Cavendish Bananas in Taiwan. Plant Dis. 1986, 70, 814–818. [Google Scholar] [CrossRef]
- Pegg, K.; Moore, N.; Bentley, S. Fusarium wilt of banana in Australia: A review. Aust. J. Agric. Res. 1996, 47, 637–650. [Google Scholar] [CrossRef]
- Ploetz, R.C. Fusarium wilt of banana is caused by several pathogens referred to as Fusarium oxysporum f. sp. cubense. Phytopathology 2006, 96, 653–656. [Google Scholar] [CrossRef] [Green Version]
- Ploetz, R.; Pegg, K. Fusarium wilt of banana and Wallace’s line: Was the disease originally restricted to his Indo-Malayan region? Australas. Plant Pathol. 1997, 26, 239–249. [Google Scholar] [CrossRef]
- Mostert, D.; Molina, A.B.; Daniells, J.; Fourie, G.; Hermanto, C.; Chao, C.-P.; Fabregar, E.; Sinohin, V.G.; Masdek, N.; Thangavelu, R.; et al. The distribution and host range of the banana Fusarium wilt fungus, Fusarium oxysporum f. sp. cubense, in Asia. PLoS ONE 2017, 12, e0181630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentley, S.; Pegg, K.; Dale, J. Genetic variation among a world-wide collection of isolates of Fusarium oxysporum f. sp. cubense analysed by RAPD-PCR fingerprinting. Mycol. Res. 1995, 99, 1378–1384. [Google Scholar] [CrossRef]
- Katan, T. Current status of vegetative compatibility groups in Fusarium oxysporum. Phytoparasitica 1999, 27, 273–277. [Google Scholar] [CrossRef]
- Correll, J.C. The Relationship between formae speciales, races and vegetative compatibility groups in Fusarium oxysporum. Phytopathology 1991, 81, 1061–1064. [Google Scholar]
- Leslie, J.F. Genetic exchange within sexual and asexual population of the genus Fusarium. In Fusarium Wilt of Banana; Ploetz, R.C., Ed.; APS Press: St. Paul, MN, USA, 1990; pp. 37–48. [Google Scholar]
- Jones, D.R. Introduction to banana, abacá and enset. In Diseases of Banana, Abaca and Enset, 1st ed.; Jones, D.R., Ed.; CABI Publishing: Droitwich Spa, UK, 2000; pp. 1–36. [Google Scholar]
- Brake, V.M.; Pegg, K.G.; Irwin, J.A.G.; Langdon, P.W. Vegetative compatibility groups within Australian populations of Fusarium oxysporum f. sp. cubense, the cause of Fusarium wilt of bananas. Aust. J. Agric. Res. 1990, 41, 863–870. [Google Scholar] [CrossRef]
- Wibowo, A.; Subandiyah, S.; Sumardiyono, C.; Sulistyowati, L.; Taylor, P.; Fegan, M. Occurrence of Tropical Race 4 of Fusarium oxysporum f. sp. cubense in Indonesia. Plant Pathol. J. 2011, 27, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Ploetz, R.C. Population biology of Fusarium oxysporum f. sp. cubense. In Fusarium Wilt of Banana; Ploetz, R.C., Ed.; APS Press: St. Paul, MN, USA, 1990; pp. 63–67. [Google Scholar]
- Koenig, R.L.; Ploetz, R.C.; Kistler, H.C. Fusarium oxysporum f. sp. cubense consists of a small number of divergent and globally distributed clonal lineages. Phytopathology 1997, 87, 915–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentley, S.; Bassam, B.J. A robust DNA amplification fingerprinting system applied to analysis of genetic variation within Fusarium oxysporum f. sp. cubense. J. Phytopathol. 1996, 144, 207–213. [Google Scholar] [CrossRef]
- Bentley, S.; Pegg, K.G.; Moore, N.Y.; Davis, R.D.; Buddenhagen, I.W. Genetic variation among vegetative compatibility groups of Fusarium oxysporum f. sp. cubense analyzed by DNA fingerprinting. Phytopathology 1998, 88, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- Groenewald, S.; Van Den Berg, N.; Marasas, W.F.; Viljoen, A. The application of high-throughput AFLP’s in assessing genetic diversity in Fusarium oxysporum f. sp. cubense. Mycol. Res. 2006, 110, 297–305. [Google Scholar] [CrossRef]
- O’Donnell, K.; Kistler, H.; Cigelnik, E.; Ploetz, R.C. Multiple evolutionary origins of the fungus causing Panama disease of banana: Concordant evidence from nuclear and mitochondrial gene genealogies. Proc. Natl. Acad. Sci. USA 1998, 95, 2044–2049. [Google Scholar] [CrossRef] [Green Version]
- Fourie, G.; Steenkamp, E.T.; Gordon, T.R.; Viljoen, A. Evolutionary Relationships among the Fusarium oxysporum f. sp. cubense vegetative compatibility groups. Appl. Environ. Microbiol. 2009, 75, 4770–4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czislowski, E.; Fraser-Smith, S.; Zander, M.; O’Neill, W.T.; Meldrum, R.A.; Tran-Nguyen, L.T.T.; Batley, J.; Aitken, E.A.B. Investigation of the diversity of effector genes in the banana pathogen, Fusarium oxysporum f. sp. cubense, reveals evidence of horizontal gene transfer. Mol. Plant Pathol. 2017, 19, 1155–1171. [Google Scholar] [CrossRef] [Green Version]
- Maryani, N.; Lombard, L.; Poerba, Y.S.; Subandiyah, S.; Crous, P.W.; Kema, G.H.J. Phylogeny and genetic diversity of the banana Fusarium wilt pathogen Fusarium oxysporum f. sp. cubense in the Indonesian centre of origin. Stud. Mycol. 2018, 92, 155–194. [Google Scholar] [CrossRef] [PubMed]
- Laurence, M.H.; Summerell, B.A.; Burgess, L.W.; Liew, E.C. Genealogical concordance phylogenetic species recognition in the Fusarium oxysporum species complex. Fungal Biol. 2014, 118, 374–384. [Google Scholar] [CrossRef]
- Sharma, M.; Nagavardhini, A.; Thudi, M.; Ghosh, R.; Pande, S.; Varshney, R.K. Development of DArT markers and assessment of diversity in Fusarium oxysporum f. sp. ciceris, wilt pathogen of chickpea (Cicer arietinum L.). BMC Genom. 2014, 15, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaccoud, D. Diversity Arrays: A solid state technology for sequence information independent genotyping. Nucleic Acids Res. 2001, 29, e25. [Google Scholar] [CrossRef] [Green Version]
- Ordonez, N.; Seidl, M.F.; Waalwijk, C.; Drenth, A.; Kilian, A.; Thomma, B.P.H.J.; Ploetz, R.C.; Kema, G.H.J. Worse comes to worst: Bananas and Panama Disease—When plant and pathogen clones meet. PLoS Pathog. 2015, 11, e1005197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.-J.; Van Der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.-J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Boehm, E.W.A.; Ploetz, R.C.; Kistler, H.C. Statistical analysis of electrophoretic karyotype variation among vegetative compatibility groups of Fusarium oxysporum f. sp. cubense. Mol. Plant-Microbe Interact. 1994, 7, 196–207. [Google Scholar] [CrossRef]
- Baayen, R.P.; O’Donnell, K.; Bonants, P.J.M.; Cigelnik, E.; Kroon, L.P.N.M.; Roebroeck, E.J.A.; Waalwijk, C. Gene genealogies and AFLP analyses in the Fusarium oxysporum complex identify monophyletic and nonmonophyletic formae speciales causing wilt and rot disease. Phytopathology 2000, 90, 891–900. [Google Scholar] [CrossRef] [Green Version]
- Solpot, T.C.; Pangga, I.B.; Baconguis, R.D.; Cumagun, C.J.R. Occurrence of Fusarium oxysporum f. sp. cubense tropical race 4 and other genotypes in Banana in South-Central Mindanao, Philippines. Philipp. Agric. Sci. 2016, 99, 370–378. [Google Scholar]
- Karangwa, P.; Mostert, D.; Ndayihanzamaso, P.; Dubois, T.; Niere, B.; Zum Felde, A.; Schouten, A.; Blomme, G.; Beed, F.; Viljoen, A. Genetic Diversity of Fusarium oxysporum f. sp. cubense in East and Central Africa. Plant Dis. 2018, 102, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Dita, M.A.; Waalwijk, C.; Buddenhagen, I.W.; Souza, M.T.S., Jr.; Kema, G.H.J. A molecular diagnostic for tropical race 4 of the banana Fusarium wilt pathogen. Plant Pathol. 2010, 59, 348–357. [Google Scholar] [CrossRef]
- Nelson, P.E.; Toussoun, T.A.; Marasas, W.F.O. Fusarium Species: An Illustrated Manual for Identification; Pennsylvania State University Press: University Park, PA, USA, 1983. [Google Scholar]
- Viljoen, A.; Mahuku, G.; Massawe, C.; Ssali, R.T.; Kimunye, J.; Mostert, G.; Ndayihanzamasu, P.; Coyne, D.L. Banana Diseases and Pests: Field Guide for Disease Diagnostics and Data Collection; International Institute of Tropical Agriculture (IITA): Ibadan, Nigeria, 2017. [Google Scholar]
- Puhalla, J.E. Classification of strains of Fusarium oxysporum on the basis of vegetative compatibility. Can. J. Bot. 1985, 63, 179–183. [Google Scholar] [CrossRef]
- O’Donnell, K.; Sutton, D.A.; Rinaldi, M.G.; Sarver, B.A.J.; Balajee, S.A.; Schroers, H.-J.; Summerbell, R.C.; Robert, V.A.R.G.; Crous, P.W.; Zhang, N.; et al. Internet-accessible DNA sequence database for identifying Fusaria from human and animal infections. J. Clin. Microbiol. 2010, 48, 3708–3718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Kuma, K.I.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Kazutoh, K.; Misakwa, K.; Kumazuharu, M.; Kei-ichi, K.; Miyatakashi, M.T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, K.; Gueidan, C.; Sink, S.; Johnston, P.R.; Crous, P.W.; Glenn, A.; Riley, R.; Zitomer, N.C.; Colyer, P.; Waalwijk, C.; et al. A two-locus DNA sequence database for typing plant and human pathogens within the Fusarium oxysporum species complex. Fungal Genet. Biol. 2009, 46, 936–948. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.; Sutton, D.A.; Rinaldi, M.G.; Magnon, K.C.; Cox, P.A.; Revankar, S.G.; Sanche, S.; Geiser, D.M.; Juba, J.H.; van Burik, J.-A.H.; et al. Genetic diversity of human pathogenic members of the Fusarium oxysporum complex inferred from multilocus DNA sequence data and amplified fragment length polymorphism analyses: Evidence for the recent dispersion of a geographically widespread clonal lineage and nosocomial origin. J. Clin. Microbiol. 2004, 42, 5109–5120. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart model selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swofford, D.L. PAUP: Phylogenetic Analysis Using Parsimony; Sinauer Associates Inc. Publishers: Sunderland, MA, USA, 1998. [Google Scholar]
- Lee, M.S.Y. Uninformative characters and apparent conflict between molecules and morphology. Mol. Biol. Evol. 2001, 18, 676–680. [Google Scholar] [CrossRef] [Green Version]
- Darlu, P.; Lecointre, G. When does the incongruence length difference test fail? Mol. Biol. Evol. 2002, 19, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, B.; Georges, A. dartR: Importing and Analysing SNP and Silicodart Data Generated by Genome-Wide Restriction Fragment Analysis. Available online: https://cran.r-project.org/web/packages/dartR/index.html (accessed on 8 September 2021).
- Gruber, B.; Unmack, P.J.; Berry, O.F.; Georges, A. dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol. Ecol. Resour. 2017, 18, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Gruber, B.; Unmack, P.J.; Berry, O.F.; Georges, A. dartR. Available online: https://cran.r-project.org/web/packages/dartR/dartR.pdf (accessed on 8 September 2021).
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2005, 23, 254–267. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [Green Version]
- Beugin, M.-P.; Gayet, T.; Pontier, D.; Devillard, S.; Jombart, T. A fast likelihood solution to the genetic clustering problem. Methods Ecol. Evol. 2017, 9, 1006–1016. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T.; Ahmed, I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Trenas, A.; Prieto, P.; Cañizares, M.C.; García-Pedrajas, M.D.; Pérez-Artés, E. Mycovirus Fusarium oxysporum f. sp. dianthi virus 1 decreases the colonizing efficiency of its fungal host. Front. Cell. Infect. Microbiol. 2019, 9, 51. [Google Scholar] [CrossRef]
- Agbetiameh, D.; Ortega-Beltran, A.; Awuah, R.T.; Atehnkeng, J.; Islam, M.S.; Callicott, K.A.; Cotty, P.J.; Bandyopadhyay, R. Potential of atoxigenic Aspergillus flavus vegetative compatibility groups associated with maize and groundnut in Ghana as biocontrol agents for aflatoxin management. Front. Microbol. 2019, 10, 2069. [Google Scholar] [CrossRef] [PubMed]
- Fourie, G.; Steenkamp, E.T.; Ploetz, R.C.; Gordon, T.R.; Viljoen, A. Current status of the taxonomic position of Fusarium oxysporum f. sp. cubense within the Fusarium oxysporum complex. Infect. Genet. Evol. 2011, 11, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.W.; Jacobson, D.J.; Fisher, M.C. The evolution of ssexual fungi: Reproduction, speciation and classification. Annu. Rev. Phytopathol. 1999, 37, 197–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buxton, E. Parasexual recombination in the banana-wilt Fusarium. Trans. Br. Mycol. Soc. 1962, 45, 274–279. [Google Scholar] [CrossRef]
- Kuhn, D.N.; Cortes, B.; Pinto, T.; Weaver, J. Parasexuality and heterokaryosis in Fusarium oxysporum f. sp. cubense. Phytopathology 1995, 85, 1119. [Google Scholar]
- Blomme, G.; Ploetz, R.C.; Jones, D.; De Langhe, E.; Price, N.; Gold, C.; Geering, A.D.; Viljoen, A.; Karamura, D.E.; Pillay, M.; et al. A historical overview of the appearance and spread of Musa pests and pathogens on the African continent: Highlighting the importance of clean Musa planting materials and quarantine measures. Ann. Appl. Biol. 2013, 162, 4–26. [Google Scholar] [CrossRef]
- Maldonado-Bonilla, L.D.; Calderón-Oropeza, M.A.; Villarruel-Ordaz, J.L.; Sãnchez-Espinosa, A.C. Identification of novel potential causal agents of Fusarium wilt of Musa sp. AAB in southern Mexico. J. Plant Pathol. Microbiol. 2019, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Drenth, A.; McTaggart, A.R.; Wingfield, B.D. Fungal clones win the battle, but recombination wins the war. IMA Fungus 2019, 10, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, T.R. Population structure and the relationship between pathogenic and non-pathogenic strains of Fusarium oxysporum. Phytopathology 1992, 82, 73–77. [Google Scholar] [CrossRef]
- Magdama, F.; Monserrate-Maggi, L.; Serrano, L.; Sosa, D.; Geiser, D.M.; del Mar Jiménez-Gasco, M.D.M. Comparative analysis uncovers the limitations of current molecular detection methods for Fusarium oxysporum f. sp. cubense race 4 strains. PLoS ONE 2019, 14, e0222727. [Google Scholar] [CrossRef]
- Ma, L.; Houterman, P.M.; Gawehns, F.; Cao, L.; Sillo, F.; Richter, H.; Clavijo-Ortiz, M.J.; Schmidt, S.M.; Boeren, S.; Vervoort, J.; et al. The AVR 2–SIX 5 gene pair is required to activate I-2-mediated immunity in tomato. New Phytol. 2015, 208, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Rep, M.; Meijer, M.; Houterman, P.M.; van der Does, H.C.; Cornelissen, B.J.C. Fusarium oxysporum cvades I-3-mediated resistance without altering the matching avirulence Gene. Mol. Plant-Microbe Interact. 2005, 18, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Rep, M.; Van Der Does, H.C.; Meijer, M.; Van Wijk, R.; Houterman, P.M.; Dekker, H.L.; De Koster, C.G.; Cornelissen, B.J.C. A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol. Microbiol. 2004, 53, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Widinugraheni, S.; Niño-Sánchez, J.; Van Der Does, H.C.; Van Dam, P.; García-Bastidas, F.A.; Subandiyah, S.; Meijer, H.J.G.; Kistler, H.C.; Kema, G.H.J.; Rep, M. A SIX1 homolog in Fusarium oxysporum f. sp. cubense tropical race 4 contributes to virulence towards Cavendish banana. PLoS ONE 2018, 13, e0205896. [Google Scholar] [CrossRef]
- Brankovics, B.; Van Dam, P.; Rep, M.; De Hoog, G.S.; Van Der Lee, T.A.J.; Waalwijk, C.; Van Diepeningen, A.D.; Brankovics, B.; Van Dam, P.; Rep, M.; et al. Mitochondrial genomes reveal recombination in the presumed asexual Fusarium oxysporum species complex. BMC Genom. 2017, 18, 735. [Google Scholar] [CrossRef] [Green Version]
- Summerell, B.A. Resolving Fusarium: Current Status of the Genus. Annu. Rev. Phytopathol. 2019, 57, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Torres-Bedoya, E.T.; Bebber, D.; Studholme, D.J. Taxonomic revision of the banana Fusarium wilt TR4 pathogen is premature. Phytopathology 2021, in press. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Clade | SC Grouping | VCGs |
|---|---|---|
| A | 1 | 0120/15, 0122, 0126, 0129/11, 01210, 01219, 01227, SMV1, SMV2 |
| 2 | 0121, 01213/16, SMV3 | |
| C | 3 | BRIP62834, 01226 |
| B | 4 | 0124/5, 0128, 01212, 01220, 01222, 01228, 01229, 01232,01233, 01236, 01237, SMV4–7, SMV13, SMV 17 |
| 5 | 01214, 01231, SMV12, BRIP62833 | |
| 6 | 0123, 01217, 01223, 01224, 01225, 01238, SMV 15–16 | |
| 7 | 01218, 01234, 01235, SMV8–11, BRIP62618, BRIP62577 | |
| 8 | 01221, 01230 |
| SIX Gene | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Culture Collection | VCG | 1a–g | 1h | 1i | 2 | 4 | 6 | 7 | 8a | 8b | 9a | 9b | 9c | 10 | 13a | 13b + c | 13d + e |
| 44,012 * | 0120 | + | - | - | + | + | - | + | + | + | + | - | - | - | - | - | - |
| 62,892 * | 0122 | + | - | + | - | - | - | - | + | - | + | - | - | - | - | + | - |
| 59,161 * | 0126 | + | - | - | + | + | - | + | + | + | + | - | - | - | - | - | - |
| 40,255 * | 0129 | + | - | - | + | + | - | + | + | + | + | - | - | - | - | - | - |
| 26,029 * | 01210 | + | - | - | + | + | + | - | - | - | + | - | - | - | + | - | - |
| 39,259 * | 01211 | + | - | - | + | + | - | + | + | + | + | - | - | - | - | - | - |
| 36,112 * | 01215 | + | - | - | + | + | - | + | + | + | + | - | - | - | - | - | - |
| 63,261 * | 01219 | + | - | - | + | + | - | + | + | + | + | - | - | - | - | - | - |
| 615 | 01227 | + | - | - | + | + | + | - | + | - | + | - | - | - | - | - | - |
| 59,028 * | 0120/15 | + | - | - | + | + | - | + | + | + | + | - | - | - | - | - | - |
| 600 | SMV 1 | + | - | - | - | + | + | + | + | + | + | - | - | - | - | - | - |
| 818 | SMV 2 | + | - | - | - | + | + | - | - | - | + | - | - | - | - | - | - |
| 62,962 * | 0121 | + | + | + | + | + | + | + | + | - | + | - | - | + | - | - | + |
| 40,340 * | 01213 | + | + | + | + | + | + | - | + | - | + | - | - | - | + | - | + |
| 59,049 * | 01216 | + | + | + | + | + | + | - | + | - | + | - | - | - | - | - | + |
| 1683 | 01213/16 | + | + | + | + | + | + | - | + | - | + | - | - | - | + | - | + |
| 2357 | 01213/16 | + | + | + | + | + | + | - | + | - | + | - | - | - | + | - | + |
| 3372 | 01213/16 | + | + | + | + | + | + | - | + | - | + | - | - | - | + | - | - |
| 616 | SMV 3 | + | - | - | + | + | + | - | + | + | + | - | - | + | - | - | - |
| 1024 | 01226 | + | - | - | - | + | + | - | - | - | + | - | - | - | - | - | - |
| 62,933 * | 0124 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 62,957 * | 0125 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 22,887 * | 0128 | + | - | - | - | + | + | - | - | - | + | - | - | - | - | - | - |
| 62,955 * | 01212 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 58,803 * | 01220 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 853 | 01222 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 59,170 * | 01222 | + | - | - | - | - | - | - | - | - | + | - | - | - | + | - | - |
| 620 | 01228 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 898 | 01229 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 1907 | 01232 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 1910 | 01232 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 1942 | 01233 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 1960 | 01233 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 1970 | 01233 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 2413 | 01236 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | + | - |
| 2407 | 01236 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | + | - |
| 3142 | 01237 | + | - | - | - | - | + | - | - | - | + | - | - | - | + | - | - |
| 3143 | 01237 | + | - | - | - | - | + | - | - | - | + | - | - | - | + | - | - |
| 58,813 * | 0124/22 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 3475 | 0124/5 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 3478 | 0124/5 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 3481 | 0124/5 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 1004 | 0128/20 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 3474 | SMV 13 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 3484 | SMV 14 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 3127 | SMV 17 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 624 | SMV 4 | + | - | - | - | - | + | - | - | - | + | - | - | - | + | - | - |
| 1927 | SMV 5 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 1933 | SMV 6 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 1974 | SMV 7 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 25,609 * | 01214 | + | - | - | - | - | - | - | - | - | + | - | + | - | + | - | - |
| 1833 | 01231 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 3128 | 01231 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 3130 | 01231 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 2611 | Non-pathogen | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| 2633 | Non-pathogen | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| 2734 | SMV 12 | + | - | - | - | - | + | - | - | - | + | - | + | - | + | - | - |
| 62,895 * | 0123 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 58,698 * | 01217 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 36,116 * | 01223 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 909 | 01225 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 3351 | 01238 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 3371 | 01238 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 3534 | SMV 15 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 3540 | SMV 16 | + | - | - | - | - | + | - | - | - | + | - | - | - | + | - | - |
| 63,259 * | 01218 | + | - | - | - | + | + | - | - | - | + | + | - | - | + | - | - |
| 2266 | 01234 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 2267 | 01234 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 2271 | SMV 10 | + | - | - | - | + | + | - | - | - | + | - | + | - | - | - | - |
| 2351 | SMV 11 | + | - | - | - | + | + | - | - | - | + | + | - | - | + | - | - |
| 2075 | SMV 8 | + | - | - | - | + | + | - | - | - | + | - | - | - | - | - | - |
| 2257 | SMV 9 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 2281 | 01235 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 2287 | 01235 | + | - | - | - | + | + | - | - | - | + | - | - | - | + | - | - |
| 1003 | 01221 | + | - | - | - | - | - | - | - | - | + | - | - | - | + | - | + |
| 36,118 * | 01221 | + | - | - | - | - | - | - | - | - | + | - | - | - | + | - | + |
| 1011 | 01230 | + | - | - | - | - | + | - | - | - | + | - | - | - | + | - | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mostert, D.; Wicker, E.; de Jager, M.M.; Al Kaabi, S.M.; O’Neill, W.T.; Perry, S.; Li, C.; Ganyun, Y.; Pegg, K.G.; Mostert, L.; et al. A Polyphasic Approach Reveals Novel Genotypes and Updates the Genetic Structure of the Banana Fusarium Wilt Pathogen. Microorganisms 2022, 10, 269. https://doi.org/10.3390/microorganisms10020269
Mostert D, Wicker E, de Jager MM, Al Kaabi SM, O’Neill WT, Perry S, Li C, Ganyun Y, Pegg KG, Mostert L, et al. A Polyphasic Approach Reveals Novel Genotypes and Updates the Genetic Structure of the Banana Fusarium Wilt Pathogen. Microorganisms. 2022; 10(2):269. https://doi.org/10.3390/microorganisms10020269
Chicago/Turabian StyleMostert, Diane, Emmanuel Wicker, Mignon M. de Jager, Saif M. Al Kaabi, Wayne T. O’Neill, Suzy Perry, Chunyu Li, Yi Ganyun, Kenneth G. Pegg, Lizel Mostert, and et al. 2022. "A Polyphasic Approach Reveals Novel Genotypes and Updates the Genetic Structure of the Banana Fusarium Wilt Pathogen" Microorganisms 10, no. 2: 269. https://doi.org/10.3390/microorganisms10020269