
Gut Microbiota, Macrophages and Diet: An Intriguing New Triangle in Intestinal Fibrosis


 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
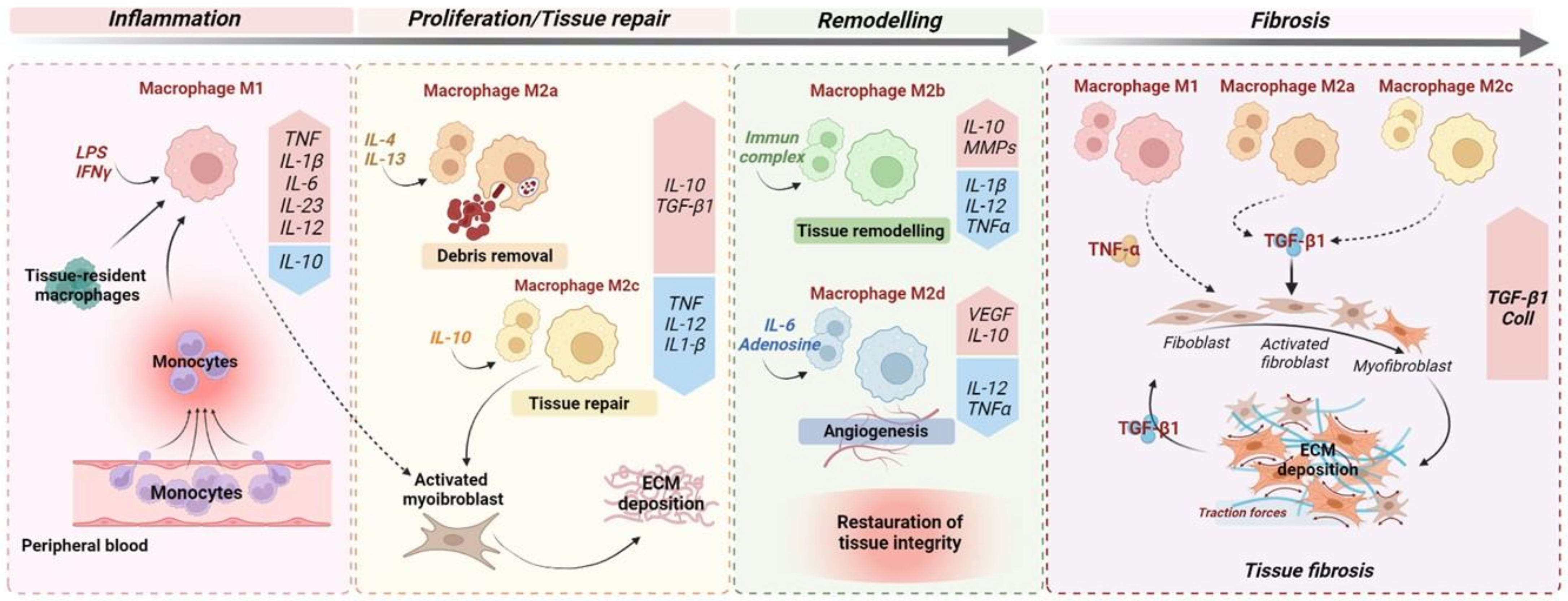
2. Macrophages as Key Players in Fibrogenesis
3. Factors That Influence Macrophage Polarisation in Inflammation and Fibrosis
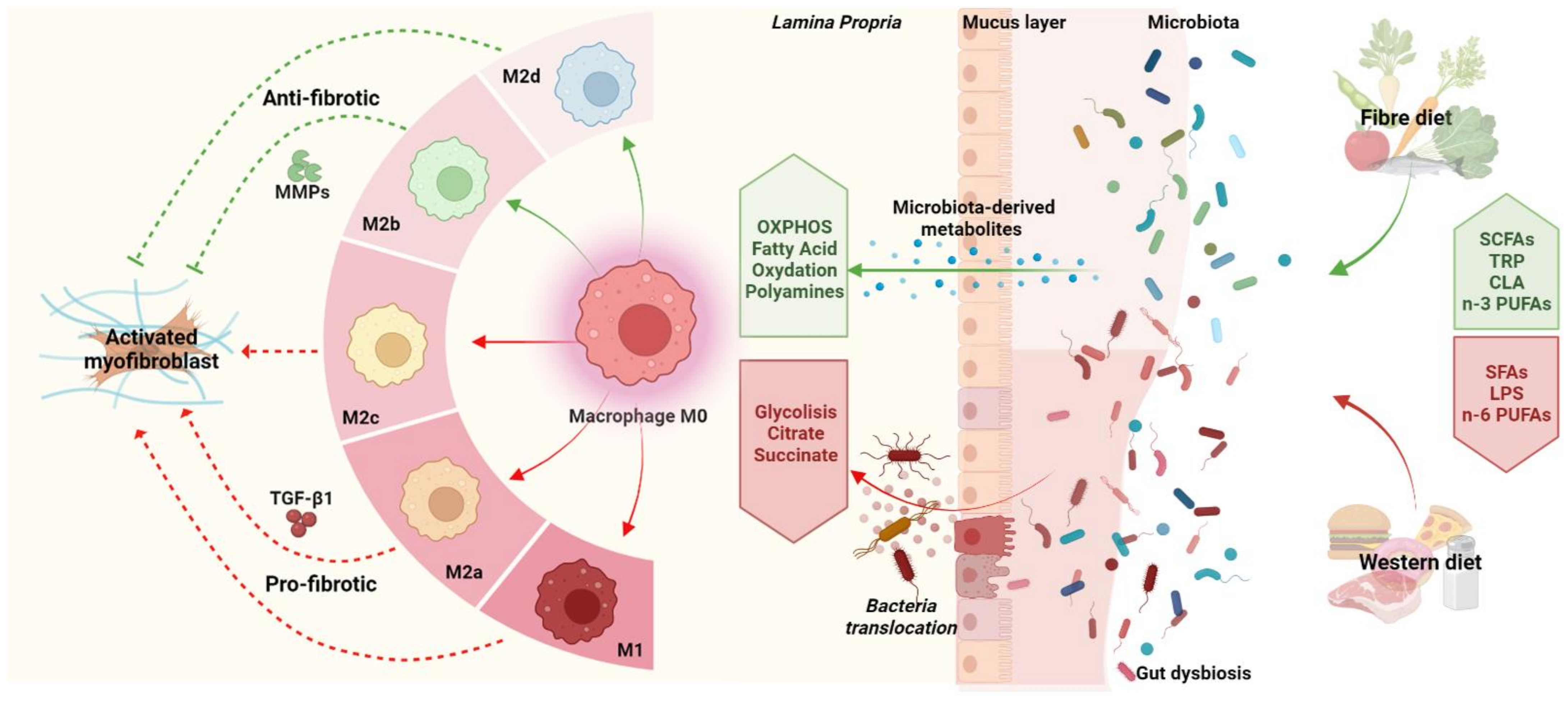
3.1. Gut Microbiota
3.2. Diet
4. Interplay between Diet, Gut Microbiota, and Macrophages in Intestinal Fibrosis
5. Reprogramming Macrophages for Treatment of Fibrosis: Future Directions
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holvoet, T.; Devriese, S.; Castermans, K.; Boland, S.; Leysen, D.; Vandewynckel, Y.-P.; Devisscher, L.; Van den Bossche, L.; Van Welden, S.; Dullaers, M.; et al. Treatment of Intestinal Fibrosis in Experimental Inflammatory Bowel Disease by the Pleiotropic Actions of a Local Rho Kinase Inhibitor. Gastroenterology 2017, 153, 1054–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieder, F. Managing Intestinal Fibrosis in Patients With Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2018, 14, 120–122. [Google Scholar]
- Lewis, R.T.; Maron, D.J. Efficacy and complications of surgery for Crohn’s disease. Gastroenterol. Hepatol. 2010, 6, 587–596. [Google Scholar]
- Andres, P.G.; Friedman, L.S. Epidemiology and the natural course of inflammatory bowel disease. Gastroenterol. Clin. N. Am. 1999, 28, 255–281. [Google Scholar] [CrossRef]
- Szigethy, E.M.; Allen, J.I.; Reiss, M.; Cohen, W.; Perera, L.P.; Brillstein, L.; Cross, R.K.; Schwartz, D.A.; Kosinski, L.R.; Colton, J.B.; et al. White Paper AGA: The Impact of Mental and Psychosocial Factors on the Care of Patients With Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. 2017, 15, 986–997. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, S.J.; Allen, J.E. The expanding world of tissue-resident macrophages. Eur. J. Immunol. 2021, 51, 1882–1896. [Google Scholar] [CrossRef]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Wang, S.Y.; Kwak, G.; Yang, Y.; Kwon, I.C.; Kim, S.H. Exosome-Guided Phenotypic Switch of M1 to M2 Macrophages for Cutaneous Wound Healing. Adv. Sci. 2019, 6, 1900513. [Google Scholar] [CrossRef] [Green Version]
- Vaday, G.G.; Lider, O. Extracellular matrix moieties, cytokines, and enzymes: Dynamic effects on immune cell behavior and inflammation. J. Leukoc. Biol. 2000, 67, 149–159. [Google Scholar] [CrossRef]
- Desmoulière, A.; Badid, C.; Mounier, N.; Costa, A.M. Role of myofibroblasts during normal tissue repair and excessive scarring: Interest of their assessment in nephropathies. Histol. Histopathol. 2000, 15, 269–280. [Google Scholar] [CrossRef]
- Kolliniati, O.; Ieronymaki, E.; Vergadi, E.; Tsatsanis, C. Metabolic Regulation of Macrophage Activation. J. Innate Immun. 2021, 14, 51–68. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Mahida, Y.R. The Key Role of Macrophages in the Immunopathogenesis of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2000, 6, 21–33. [Google Scholar] [CrossRef]
- Wang, J.; Chen, W.-D.; Wang, Y.-D. The Relationship Between Gut Microbiota and Inflammatory Diseases: The Role of Macrophages. Front. Microbiol. 2020, 11, 1065. [Google Scholar] [CrossRef]
- Lopes, T.C.M.; Mosser, D.M.; Gonçalves, R. Macrophage polarization in intestinal inflammation and gut homeostasis. Agents Actions 2020, 69, 1163–1172. [Google Scholar] [CrossRef]
- Grimm, M.C.; Pullman, W.E.; Bennett, G.M.; Sullivan, P.J.; Pavli, P.; Doe, W.F. Direct evidence of monocyte recruitment to inflammatory bowel disease mucosa. J. Gastroenterol. Hepatol. 1995, 10, 387–395. [Google Scholar] [CrossRef]
- Xiao, X.; Gaffar, I.; Guo, P.; Wiersch, J.; Fischbach, S.; Peirish, L.; Song, Z.; El-Gohary, Y.; Prasadan, K.; Shiota, C.; et al. M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc. Natl. Acad. Sci. USA 2014, 111, E1211–E1220. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Gon, Y.; Takeshita, I.; Maruoka, S.; Horie, T. IL-4 and IL-13 induce myofibroblastic phenotype of human lung fibroblasts through c-Jun NH2-terminal kinase–dependent pathway. J. Allergy Clin. Immunol. 2001, 107, 1001–1008. [Google Scholar] [CrossRef]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Kipari, T.; Haslett, C.; Iredale, J.P.; Liu, F.-T.; Hughes, J.; Sethi, T. Galectin-3 Expression and Secretion Links Macrophages to the Promotion of Renal Fibrosis. Am. J. Pathol. 2008, 172, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Shi, J.; Chen, L.; Lv, Z.; Chen, X.; Cao, H.; Xiang, Z.; Han, X. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun. Signal. 2018, 16, 12. [Google Scholar] [CrossRef] [Green Version]
- Nikolic-Paterson, D.; Wang, S.; Lan, H.Y. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. Suppl. 2014, 4, 34–38. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; You, H.; Fan, X.; Jia, J. Hepatic macrophages in liver fibrosis: Pathogenesis and potential therapeutic targets. BMJ Open Gastroenterol. 2016, 3, e000079. [Google Scholar] [CrossRef]
- Lenti, M.V.; Di Sabatino, A. Intestinal fibrosis. Mol. Asp. Med. 2018, 65, 100–109. [Google Scholar] [CrossRef]
- Hu, H.-H.; Chen, D.-Q.; Wang, Y.-N.; Feng, Y.-L.; Cao, G.; Vaziri, N.D.; Zhao, Y.-Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Rieder, F.; Kessler, S.P.; West, G.A.; Bhilocha, S.; de la Motte, C.; Sadler, T.M.; Gopalan, B.; Stylianou, E.; Fiocchi, C. Inflammation-Induced Endothelial-to-Mesenchymal Transition: A Novel Mechanism of Intestinal Fibrosis. Am. J. Pathol. 2011, 179, 2660–2673. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Castaño, A.P.; Nowlin, B.T.; Lupher, M.L.; Duffield, J.S. Bone Marrow Ly6Chigh Monocytes Are Selectively Recruited to Injured Kidney and Differentiate into Functionally Distinct Populations. J. Immunol. 2009, 183, 6733–6743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.G.; Homer, R.; Zhu, Z.; Lanone, S.; Wang, X.; Koteliansky, V.E.; Shipley, J.M.; Gotwals, P.J.; Noble, P.W.; Chen, Q.; et al. Interleukin-13 Induces Tissue Fibrosis by Selectively Stimulating and Activating Transforming Growth Factor β1. J. Exp. Med. 2001, 194, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Ide, M.; Kuwamura, M.; Kotani, T.; Sawamoto, O.; Yamate, J. Effects of Gadolinium Chloride (GdCl3) on the Appearance of Macrophage Populations and Fibrogenesis in Thioacetamide-Induced Rat Hepatic Lesions. J. Comp. Pathol. 2005, 133, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Carlson, S.; Helterline, D.; Asbe, L.; Dupras, S.; Minami, E.; Farris, S.; Stempien-Otero, A. Cardiac macrophages adopt profibrotic/M2 phenotype in infarcted hearts: Role of urokinase plasminogen activator. J. Mol. Cell. Cardiol. 2016, 108, 42–49. [Google Scholar] [CrossRef]
- Shen, B.; Liu, X.; Fan, Y.; Qiu, J. Macrophages Regulate Renal Fibrosis Through Modulating TGFβ Superfamily Signaling. Inflammation 2014, 37, 2076–2084. [Google Scholar] [CrossRef]
- Ferrante, C.J.; Leibovich, S.J. Regulation of Macrophage Polarization and Wound Healing. Adv. Wound Care 2012, 1, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Tseng, W.-C.; Tsai, M.-T.; Chen, N.-J.; Tarng, D.-C. Trichostatin A Alleviates Renal Interstitial Fibrosis Through Modulation of the M2 Macrophage Subpopulation. Int. J. Mol. Sci. 2020, 21, 5966. [Google Scholar] [CrossRef]
- Lawrance, I.C.; Rogler, G.; Bamias, G.; Breynaert, C.; Florholmen, J.; Pellino, G.; Reif, S.; Speca, S.; Latella, G. Cellular and Molecular Mediators of Intestinal Fibrosis. J. Crohn’s Colitis 2015, 11, 1491–1503. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Huang, S.; Wang, L.; Wu, Z.; Liang, M.; Li, H.; Lv, L.; Li, W.; Wu, Z. M2b Macrophages Regulate Cardiac Fibroblast Activation and Alleviate Cardiac Fibrosis After Reperfusion Injury. Circ. J. 2020, 84, 626–635. [Google Scholar] [CrossRef]
- Lourenssen, S.R.; Blennerhassett, M.G. M2 Macrophages and Phenotypic Modulation of Intestinal Smooth Muscle Cells Characterize Inflammatory Stricture Formation in Rats. Am. J. Pathol. 2020, 190, 1843–1858. [Google Scholar] [CrossRef]
- Kono, Y.; Miyoshi, S.; Fujita, T. Dextran sodium sulfate alters cytokine production in macrophages in vitro. Pharmazie 2016, 71, 619–624. [Google Scholar] [CrossRef]
- Yang, R.; Liao, Y.; Wang, L.; He, P.; Hu, Y.; Yuan, D.; Wu, Z.; Sun, X. Exosomes Derived From M2b Macrophages Attenuate DSS-Induced Colitis. Front. Immunol. 2019, 10, 2346. [Google Scholar] [CrossRef] [Green Version]
- Kühl, A.A.; Erben, U.; Kredel, L.I.; Siegmund, B. Diversity of Intestinal Macrophages in Inflammatory Bowel Diseases. Front. Immunol. 2015, 6, 613. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Mezouar, S.; Chantran, Y.; Michel, J.; Fabre, A.; Dubus, J.-C.; Leone, M.; Sereme, Y.; Mège, J.-L.; Ranque, S.; Desnues, B.; et al. Microbiome and the immune system: From a healthy steady-state to allergy associated disruption. Hum. Microbiome J. 2018, 10, 11–20. [Google Scholar] [CrossRef]
- Ruder, B.; Becker, C. At the Forefront of the Mucosal Barrier: The Role of Macrophages in the Intestine. Cells 2020, 9, 2162. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Honda, K. Intestinal Commensal Microbes as Immune Modulators. Cell Host Microbe 2012, 12, 496–508. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, X.; Huycke, T.; Moore, D.R.; Lightfoot, S.A.; Huycke, M.M. Colon Macrophages Polarized by Commensal Bacteria Cause Colitis and Cancer through the Bystander Effect. Transl. Oncol. 2013, 6, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, A.; Sato, T.; Kamada, N.; Mikami, Y.; Matsuoka, K.; Hisamatsu, T.; Hibi, T.; Roers, A.; Yagita, H.; Ohteki, T.; et al. A Single Strain of Clostridium butyricum Induces Intestinal IL-10-Producing Macrophages to Suppress Acute Experimental Colitis in Mice. Cell Host Microbe 2013, 13, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Dauphinee, S.M.; Karsan, A. Lipopolysaccharide signaling in endothelial cells. Lab. Investig. 2005, 86, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Ha, C.W.; Martin, A.; Sepich-Poore, G.D.; Shi, B.; Wang, Y.; Gouin, K.; Humphrey, G.; Sanders, K.; Ratnayake, Y.; Chan, K.S.; et al. Translocation of Viable Gut Microbiota to Mesenteric Adipose Drives Formation of Creeping Fat in Humans. Cell 2020, 183, 666–683.e17. [Google Scholar] [CrossRef]
- Schaedler, R.W.; Dubos, R.; Costello, R. The development of the bacterial flora in the gastrointestinal tract of mice. J. Exp. Med. 1965, 122, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, Q.; Wu, J.; Wu, Y.; Peng, W.; Li, H.; Wang, J.; Tang, X.; Peng, Y.; Fu, X. Fusobacterium nucleatum promotes M2 polarization of macrophages in the microenvironment of colorectal tumours via a TLR4-dependent mechanism. Cancer Immunol. Immunother. 2018, 67, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Glasser, A.-L.; Boudeau, J.; Barnich, N.; Perruchot, M.-H.; Colombel, J.-F.; Darfeuille-Michaud, A. Adherent Invasive Escherichia coli Strains from Patients with Crohn’s Disease Survive and Replicate within Macrophages without Inducing Host Cell Death. Infect. Immun. 2001, 69, 5529–5537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chervy, M.; Barnich, N.; Denizot, J. Adherent-Invasive E. coli: Update on the Lifestyle of a Troublemaker in Crohn’s Disease. Int. J. Mol. Sci. 2020, 21, 3734. [Google Scholar] [CrossRef]
- Imai, J.; Kitamoto, S.; Sugihara, K.; Nagao-Kitamoto, H.; Hayashi, A.; Morhardt, T.L.; Kuffa, P.; Higgins, P.D.R.; Barnich, N.; Kamada, N. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. 2019, 12, 632–643. [Google Scholar] [CrossRef]
- Lacavé-Lapalun, J.-V.; Benderitter, M.; Linard, C. Flagellin or Lipopolysaccharide Treatment Modified Macrophage Populations after Colorectal Radiation of Rats. J. Pharmacol. Exp. Ther. 2013, 346, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Neely, C.J.; Kartchner, L.B.; Mendoza, A.E.; Linz, B.M.; Frelinger, J.A.; Wolfgang, M.C.; Maile, R.; Cairns, B.A. Flagellin Treatment Prevents Increased Susceptibility to Systemic Bacterial Infection after Injury by Inhibiting Anti-Inflammatory IL-10+ IL-12- Neutrophil Polarization. PLoS ONE 2014, 9, e85623. [Google Scholar] [CrossRef]
- Dall’Asta, M.; Derlindati, E.; Ardigò, D.; Zavaroni, I.; Brighenti, F.; Del Rio, D. Macrophage polarization: The answer to the diet/inflammation conundrum? Nutr. Metab. Cardiovasc. Dis. 2012, 22, 387–392. [Google Scholar] [CrossRef]
- Toomey, S.; Harhen, B.; Roche, H.; Fitzgerald, D.; Belton, O. Profound resolution of early atherosclerosis with conjugated linoleic acid. Atherosclerosis 2006, 187, 40–49. [Google Scholar] [CrossRef]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef] [Green Version]
- Marion-Letellier, R.; Dechelotte, P.; Iacucci, M.; Ghosh, S. Dietary modulation of peroxisome proliferator-activated receptor gamma. Gut 2008, 58, 586–593. [Google Scholar] [CrossRef]
- Gao, S.; Zhou, J.; Liu, N.; Wang, L.; Gao, Q.; Wu, Y.; Zhao, Q.; Liu, P.; Wang, S.; Liu, Y.; et al. Curcumin induces M2 macrophage polarization by secretion IL-4 and/or IL-13. J. Mol. Cell. Cardiol. 2015, 85, 131–139. [Google Scholar] [CrossRef]
- Saidi, A.; Kasabova, M.; Vanderlynden, L.; Wartenberg, M.; Kara-Ali, G.H.; Marc, D.; Lecaille, F.; Lalmanach, G. Curcumin inhibits the TGF-β1-dependent differentiation of lung fibroblasts via PPARγ-driven upregulation of cathepsins B and L. Sci. Rep. 2019, 9, 491. [Google Scholar] [CrossRef]
- Xu, S.; Jiang, B.; Wang, H.; Shen, C.; Chen, H.; Zeng, L. Curcumin Suppresses Intestinal Fibrosis by Inhibition of PPARγ-Mediated Epithelial-Mesenchymal Transition. Evid.-Based Complement. Altern. Med. 2017, 2017, 8140962. [Google Scholar] [CrossRef] [Green Version]
- Severo, J.S.; Barros, V.J.D.S.; da Silva, A.C.A.; Parente, J.M.L.; Lima, M.M.; Lima, A.M.; dos Santos, A.A.; Neto, E.M.M.; Tolentino, M. Effects of glutamine supplementation on inflammatory bowel disease: A systematic review of clinical trials. Clin. Nutr. ESPEN 2021, 42, 53–60. [Google Scholar] [CrossRef]
- Ryan, D.G.; O’Neill, L. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. 2017, 591, 2992–3006. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, N.; Chand, L.; Han, M.K.; Lee, S.O.; Kim, C.Y.; Jeong, Y.J. Glutamine inhibits CCl4 induced liver fibrosis in mice and TGF-β1 mediated epithelial–mesenchymal transition in mouse hepatocytes. Food Chem. Toxicol. 2016, 93, 129–137. [Google Scholar] [CrossRef]
- San-Miguel, B.; Crespo, I.; Kretzmann, N.A.; Mauriz, J.L.; Marroni, N.; Tuñón, M.J.; González-Gallego, J. Glutamine Prevents Fibrosis Development in Rats with Colitis Induced by 2,4,6-Trinitrobenzene Sulfonic Acid. J. Nutr. 2010, 140, 1065–1071. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; O’Leary, E.M.; Witt, L.J.; Tian, Y.; Gökalp, G.A.; Meliton, A.Y.; Dulin, N.O.; Mutlu, G.M. Glutamine Metabolism Is Required for Collagen Protein Synthesis in Lung Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 61, 597–606. [Google Scholar] [CrossRef]
- Yao, Q.; Li, S.; Li, X.; Wang, F.; Tu, C. Myricetin Modulates Macrophage Polarization and Mitigates Liver Inflammation and Fibrosis in a Murine Model of Nonalcoholic Steatohepatitis. Front. Med. 2020, 7, 71. [Google Scholar] [CrossRef] [Green Version]
- Lefèvre, L.; Galès, A.; Olagnier, D.; Bernad, J.; Perez, L.; Burcelin, R.; Valentin, A.; Auwerx, J.; Pipy, B.; Coste, A. PPARγ Ligands Switched High Fat Diet-Induced Macrophage M2b Polarization toward M2a Thereby Improving Intestinal Candida Elimination. PLoS ONE 2010, 5, e12828. [Google Scholar] [CrossRef]
- Bourlier, V.; Zakaroff-Girard, A.; Miranville, A.; De Barros, S.; Maumus, M.; Sengenes, C.; Galitzky, J.; Lafontan, M.; Karpe, F.; Frayn, K.N.; et al. Remodeling Phenotype of Human Subcutaneous Adipose Tissue Macrophages. Circulation 2008, 117, 806–815. [Google Scholar] [CrossRef]
- Li, X.; Wei, X.; Sun, Y.; Du, J.; Li, X.; Xun, Z.; Li, Y.C. High-fat diet promotes experimental colitis by inducing oxidative stress in the colon. Am. J. Physiol. Liver Physiol. 2019, 317, G453–G462. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Konijeti, G.G.; Higuchi, L.M.; De Silva, P.; Fuchs, C.S.; Willett, W.C.; Richter, J.M.; Chan, A.T. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut 2013, 63, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Scaioli, E.; Liverani, E.; Belluzzi, A. The Imbalance between n-6/n-3 Polyunsaturated Fatty Acids and Inflammatory Bowel Disease: A Comprehensive Review and Future Therapeutic Perspectives. Int. J. Mol. Sci. 2017, 18, 2619. [Google Scholar] [CrossRef] [Green Version]
- Marion-Letellier, R.; Savoye, G.; Beck, P.L.; Panaccione, R.; Ghosh, S. Polyunsaturated Fatty Acids in Inflammatory Bowel Diseases: A reappraisal of effects and therapeutic approaches. Inflamm. Bowel Dis. 2013, 19, 650–661. [Google Scholar] [CrossRef]
- Song, M.-Y.; Wang, J.; Lee, Y.; Lee, J.; Kwon, K.-S.; Bae, E.J.; Park, B.-H. Enhanced M2 macrophage polarization in high n-3 polyunsaturated fatty acid transgenic mice fed a high-fat diet. Mol. Nutr. Food Res. 2016, 60, 2481–2492. [Google Scholar] [CrossRef]
- Tang, H.; Zhu, X.; Gong, C.; Liu, H.; Liu, F. Protective effects and mechanisms of omega-3 polyunsaturated fatty acid on intestinal injury and macrophage polarization in peritoneal dialysis rats. Nephrology 2019, 24, 1081–1089. [Google Scholar] [CrossRef] [Green Version]
- Binger, K.J.; Gebhardt, M.; Heinig, M.; Rintisch, C.; Schroeder, A.; Neuhofer, W.; Hilgers, K.; Manzel, A.; Schwartz, C.; Kleinewietfeld, M.; et al. High salt reduces the activation of IL-4– and IL-13–stimulated macrophages. J. Clin. Investig. 2015, 125, 4223–4238. [Google Scholar] [CrossRef]
- Lu, J.; Bai, Z.; Kuang, X.; Li, L. High-salt exposure induces macrophage polarization to promote proliferation and phenotypic transformation of co-cultured renal fibroblasts. Nan Fang Yi Ke Da Xue Xue Bao (J. South. Med. Univ.) 2020, 40, 1472–1479. [Google Scholar] [CrossRef]
- Preda, C.M.; Manuc, T.; Chifulescu, A.; Istratescu, D.; Louis, E.; Baicus, C.; Sandra, I.; Diculescu, M.-M.; Reenaers, C.; Van Kemseke, C.; et al. Diet as an environmental trigger in inflammatory bowel disease: A retrospective comparative study in two European cohorts. Rev. Esp. Enferm. Dig. 2020, 112, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, S.L.F.; Miranda, M.C.G.; Guimarães, M.A.F.; Santiago, H.C.; Queiroz, C.P.; Cunha, P.D.S.; Cara, D.C.; Foureaux, G.; Ferreira, A.J.; Cardoso, V.N.; et al. High-Salt Diet Induces IL-17-Dependent Gut Inflammation and Exacerbates Colitis in Mice. Front. Immunol. 2018, 8, 1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amamou, A.; Rouland, M.; Yaker, L.; Goichon, A.; Guérin, C.; Aziz, M.; Savoye, G.; Marion-Letellier, R. Dietary salt exacerbates intestinal fibrosis in chronic TNBS colitis via fibroblasts activation. Sci. Rep. 2021, 11, 15055. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef] [Green Version]
- Michaudel, C.; Sokol, H. The Gut Microbiota at the Service of Immunometabolism. Cell Metab. 2020, 32, 514–523. [Google Scholar] [CrossRef]
- Makki, K.; Deehan, E.C.; Walter, J.; Bäckhed, F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Shu, D.; Zheng, M.; Wang, J.; Luo, C.; Wang, Y.; Guo, F.; Zou, X.; Lv, X.; Li, Y.; et al. Microbial metabolite butyrate facilitates M2 macrophage polarization and function. Sci. Rep. 2016, 6, 24838. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Fava, F. Intestinal microbiota in inflammatory bowel disease: Friend of foe? World J. Gastroenterol. 2011, 17, 557–566. [Google Scholar] [CrossRef]
- Gasaly, N.; Hermoso, M.A.; Gotteland, M. Butyrate and the Fine-Tuning of Colonic Homeostasis: Implication for Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 3061. [Google Scholar] [CrossRef]
- Lee, J.Y.; Plakidas, A.; Lee, W.H.; Heikkinen, A.; Chanmugam, P.; Bray, G.; Hwang, D.H. Differential modulation of Toll-like receptors by fatty acids: Preferential inhibition by n-3 polyunsaturated fatty acids. J. Lipid Res. 2003, 44, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Hanayama, M.; Yamamoto, Y.; Utsunomiya, H.; Yoshida, O.; Liu, S.; Mogi, M.; Matsuura, B.; Takeshita, E.; Ikeda, Y.; Hiasa, Y. The mechanism of increased intestinal palmitic acid absorption and its impact on hepatic stellate cell activation in nonalcoholic steatohepatitis. Sci. Rep. 2021, 11, 13380. [Google Scholar] [CrossRef]
- Forbes, A.; Escher, J.; Hébuterne, X.; Kłęk, S.; Krznaric, Z.; Schneider, S.; Shamir, R.; Stardelova, K.; Wierdsma, N.; Wiskin, A.E.; et al. ESPEN guideline: Clinical nutrition in inflammatory bowel disease. Clin. Nutr. 2016, 36, 321–347. [Google Scholar] [CrossRef] [Green Version]
- Casanova, M.J.; Chaparro, M.; Molina, B.; Merino, O.; Batanero, R.; Dueñas-Sadornil, C.; Robledo, P.; Garcia-Albert, A.M.; Gómez-Sánchez, M.B.; Calvet, X.; et al. Prevalence of Malnutrition and Nutritional Characteristics of Patients With Inflammatory Bowel Disease. J. Crohn’s Colitis 2017, 11, 1430–1439. [Google Scholar] [CrossRef]
- Siva, S.; Rubin, D.T.; Gulotta, G.; Wroblewski, K.; Pekow, J. Zinc Deficiency is Associated with Poor Clinical Outcomes in Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2017, 23, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Skalny, A.V.; Aschner, M.; Lei, X.G.; Gritsenko, V.A.; Santamaria, A.; Alekseenko, S.I.; Prakash, N.T.; Chang, J.-S.; Sizova, E.A.; Chao, J.C.J.; et al. Gut Microbiota as a Mediator of Essential and Toxic Effects of Zinc in the Intestines and Other Tissues. Int. J. Mol. Sci. 2021, 22, 13074. [Google Scholar] [CrossRef]
- Gordon, S.R.; Vaishnava, S. Zinc supplementation modulates T helper 17 cells via the gut microbiome. J. Immunol. 2019, 202, 191.13. [Google Scholar]
- Higashimura, Y.; Takagi, T.; Naito, Y.; Uchiyama, K.; Mizushima, K.; Tanaka, M.; Hamaguchi, M.; Itoh, Y. Zinc Deficiency Activates the IL-23/Th17 Axis to Aggravate Experimental Colitis in Mice. J. Crohn’s Colitis 2019, 14, 856–866. [Google Scholar] [CrossRef]
- Xie, C.; Wan, L.; Li, C.; Feng, Y.; Kang, Y.J. Selective suppression of M1 macrophages is involved in zinc inhibition of liver fibrosis in mice. J. Nutr. Biochem. 2021, 97, 108802. [Google Scholar] [CrossRef]
- Sirisinha, S. The pleiotropic role of vitamin A in regulating mucosal immunity. Asian Pac. J. Allergy Immunol. 2015, 33, 71–89. [Google Scholar]
- Belda, E.; Voland, L.; Tremaroli, V.; Falony, G.; Adriouch, S.; Assmann, K.E.; Prifiti, E.; Aron-Wisnewsky, J.; Debédat, J.; Le Roy, T.; et al. Impairment of gut microbial biotin metabolism and host biotin status in severe obesity: Effect of biotin and prebiotic supplementation on improved metabolism. Gut 2022. [Google Scholar] [CrossRef]
- Fletcher, J.; Cooper, S.C.; Ghosh, S.; Hewison, M. The Role of Vitamin D in Inflammatory Bowel Disease: Mechanism to Management. Nutrients 2019, 11, 1019. [Google Scholar] [CrossRef] [Green Version]
- Pincikova, T.; Paquin-Proulx, D.; Sandberg, J.K.; Flodström-Tullberg, M.; Hjelte, L. Vitamin D treatment modulates immune activation in cystic fibrosis. Clin. Exp. Immunol. 2017, 189, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Sauder, K.L.; Rodansky, E.S.; Simpson, R.U.; Higgins, P.D. CARD-024, a vitamin D analog, attenuates the pro-fibrotic response to substrate stiffness in colonic myofibroblasts. Exp. Mol. Pathol. 2012, 93, 91–98. [Google Scholar] [CrossRef]
- Russo, E.; Giudici, F.; Fiorindi, C.; Ficari, F.; Scaringi, S.; Amedei, A. Immunomodulating Activity and Therapeutic Effects of Short Chain Fatty Acids and Tryptophan Post-biotics in Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 2754. [Google Scholar] [CrossRef] [Green Version]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Häsler, R.; et al. Increased Tryptophan Metabolism Is Associated With Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516.e2. [Google Scholar] [CrossRef] [Green Version]
- Manzella, C.R.; Jayawardena, D.; Pagani, W.; Li, Y.; Alrefai, W.A.; Bauer, J.; Jung, B.; Weber, C.R.; Gill, R.K. Serum Serotonin Differentiates Between Disease Activity States in Crohn’s Patients. Inflamm. Bowel Dis. 2020, 26, 1607–1618. [Google Scholar] [CrossRef]
- Rogala, A.R.; Oka, A.; Sartor, R.B. Strategies to Dissect Host-Microbial Immune Interactions That Determine Mucosal Homeostasis vs. Intestinal Inflammation in Gnotobiotic Mice. Front. Immunol. 2020, 11, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashita, C.; Hoshi, M.; Hirata, A.; Nakamoto, K.; Ando, T.; Hattori, T.; Yamamoto, Y.; Tezuka, H.; Tomita, H.; Hara, A.; et al. Kynurenine plays an immunosuppressive role in 2,4,6-trinitrobenzene sulfate-induced colitis in mice. World J. Gastroenterol. 2020, 26, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, I.; Zorzi, F.; Marafini, I.; Di Fusco, D.; Dinallo, V.; Caruso, R.; Izzo, R.; Franzè, E.; Colantoni, A.; Pallone, F.; et al. Aryl hydrocarbon receptor-driven signals inhibit collagen synthesis in the gut. Eur. J. Immunol. 2016, 46, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Pellizzari, G.; Hoskin, C.; Crescioli, S.; Mele, S.; Gotovina, J.; Chiaruttini, G.; Bianchini, R.; Ilieva, K.; Bax, H.J.; Papa, S.; et al. IgE re-programs alternatively-activated human macrophages towards pro-inflammatory anti-tumoural states. EBioMedicine 2019, 43, 67–81. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lin, Y.-X.; Qiao, S.-L.; An, H.-W.; Ma, Y.; Qiao, Z.-Y.; Rajapaksha, R.Y.J.; Wang, H. Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment. Biomaterials 2017, 112, 153–163. [Google Scholar] [CrossRef]
- Shields, C.W.; Evans, M.A.; Wang, L.L.-W.; Baugh, N.; Iyer, S.; Wu, D.; Zhao, Z.; Pusuluri, A.; Ukidve, A.; Pan, D.C.; et al. Cellular backpacks for macrophage immunotherapy. Sci. Adv. 2020, 6, eaaz6579. [Google Scholar] [CrossRef]
- Zhao, J.; Okamoto, Y.; Asano, Y.; Ishimaru, K.; Aki, S.; Yoshioka, K.; Takuwa, N.; Wada, T.; Inagaki, Y.; Takahashi, C.; et al. Sphingosine-1-phosphate receptor-2 facilitates pulmonary fibrosis through potentiating IL-13 pathway in macrophages. PLoS ONE 2018, 13, e0197604. [Google Scholar] [CrossRef] [Green Version]
- Sandborn, W.J.; Feagan, B.G.; D’Haens, G.; Wolf, D.C.; Jovanovic, I.; Hanauer, S.B.; Ghosh, S.; Petersen, A.; Hua, S.Y.; Lee, J.H.; et al. Ozanimod as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2021, 385, 1280–1291. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Martín, R.; Miquel, S.; Chain, F.; Natividad, J.M.; Jury, J.; Lu, J.; Sokol, H.; Theodorou, V.; Bercik, P.; Verdu, E.F.; et al. Faecalibacterium prausnitzii prevents physiological damages in a chronic low-grade inflammation murine model. BMC Microbiol. 2015, 15, 67. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.; Chain, F.; Miquel, S.; Lu, J.; Gratadoux, J.-J.; Sokol, H.; Verdu, E.F.; Bercik, P.; Humaran, L.G.B.; Langella, P. The Commensal Bacterium Faecalibacterium prausnitzii Is Protective in DNBS-induced Chronic Moderate and Severe Colitis Models. Inflamm. Bowel Dis. 2014, 20, 417–430. [Google Scholar] [CrossRef]
- Xu, J.; Liang, R.; Zhang, W.; Tian, K.; Li, J.; Chen, X.; Yu, T.; Chen, Q. Faecalibacterium prausnitzii-derived microbial anti-inflammatory molecule regulates intestinal integrity in diabetes mellitus mice via modulating tight junction protein expression. J. Diabetes 2019, 12, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Lenoir, M.; Martín, R.; Torres-Maravilla, E.; Chadi, S.; González-Dávila, P.; Sokol, H.; Langella, P.; Chain, F.; Bermúdez-Humarán, L.G. Butyrate mediates anti-inflammatory effects of Faecalibacterium prausnitzii in intestinal epithelial cells through Dact3. Gut Microbes 2020, 12, 1826748. [Google Scholar] [CrossRef]
- Ortiz-Masià, D.; Salvador, P.; Macias-Ceja, D.C.; Gisbert-Ferrándiz, L.; Esplugues, J.V.; Manyé, J.; Alós, R.; Navarro-Vicente, F.; Mamie, C.; Scharl, M.; et al. WNT2b Activates Epithelial-mesenchymal Transition Through FZD4: Relevance in Penetrating Crohn’s Disease. J. Crohn’s Colitis 2019, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Cosin-Roger, J.; Ortiz-Masià, M.D.; Barrachina, M.D. Macrophages as an Emerging Source of Wnt Ligands: Relevance in Mucosal Integrity. Front. Immunol. 2019, 10, 2297. [Google Scholar] [CrossRef] [PubMed]
- Roger, J.C.; Ortiz-Masia, M.D.; Calatayud, S.; Hernandez, C.; Álvarez, A.; Hinojosa, J.; Esplugues, J.V.; Barrachina, M.D. M2 Macrophages Activate WNT Signaling Pathway in Epithelial Cells: Relevance in Ulcerative Colitis. PLoS ONE 2013, 8, e78128. [Google Scholar] [CrossRef]
- Park, J.-S.; Choi, J.; Kwon, J.Y.; Jung, K.-A.; Yang, C.W.; Park, S.-H.; Cho, M.-L. A probiotic complex, rosavin, zinc, and prebiotics ameliorate intestinal inflammation in an acute colitis mouse model. J. Transl. Med. 2018, 16, 37. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amamou, A.; O’Mahony, C.; Leboutte, M.; Savoye, G.; Ghosh, S.; Marion-Letellier, R. Gut Microbiota, Macrophages and Diet: An Intriguing New Triangle in Intestinal Fibrosis. Microorganisms 2022, 10, 490. https://doi.org/10.3390/microorganisms10030490
Amamou A, O’Mahony C, Leboutte M, Savoye G, Ghosh S, Marion-Letellier R. Gut Microbiota, Macrophages and Diet: An Intriguing New Triangle in Intestinal Fibrosis. Microorganisms. 2022; 10(3):490. https://doi.org/10.3390/microorganisms10030490
Chicago/Turabian StyleAmamou, Asma, Cian O’Mahony, Mathilde Leboutte, Guillaume Savoye, Subrata Ghosh, and Rachel Marion-Letellier. 2022. "Gut Microbiota, Macrophages and Diet: An Intriguing New Triangle in Intestinal Fibrosis" Microorganisms 10, no. 3: 490. https://doi.org/10.3390/microorganisms10030490
APA StyleAmamou, A., O’Mahony, C., Leboutte, M., Savoye, G., Ghosh, S., & Marion-Letellier, R. (2022). Gut Microbiota, Macrophages and Diet: An Intriguing New Triangle in Intestinal Fibrosis. Microorganisms, 10(3), 490. https://doi.org/10.3390/microorganisms10030490