
Whole-Genome Sequencing and Comparative Genomic Analysis of Antimicrobial Producing Streptococcus lutetiensis from the Rumen

 ,
,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Isolation, Growth Conditions, and Maintenance of Microorganisms
2.2. Spectrum of Activity
2.3. Genomic DNA Extraction and 16 rRNA Gene Sequencing
2.4. Genome Sequencing and Assembly
2.5. Annotation and Comparative Genomic Analysis
2.6. CRISPR/Cas9 Characterization
2.7. Genome Mining of Biosynthetic Gene Clusters
2.8. Characterization of Bacteriocin Precursor Peptides
2.9. Ruminal Genomic Occurrence of Bacteriocin Precursor Peptides
2.10. Bacteriocin Extraction, Purification and Activity
3. Results
3.1. Isolation and Characterization of Ruminal Streptococci
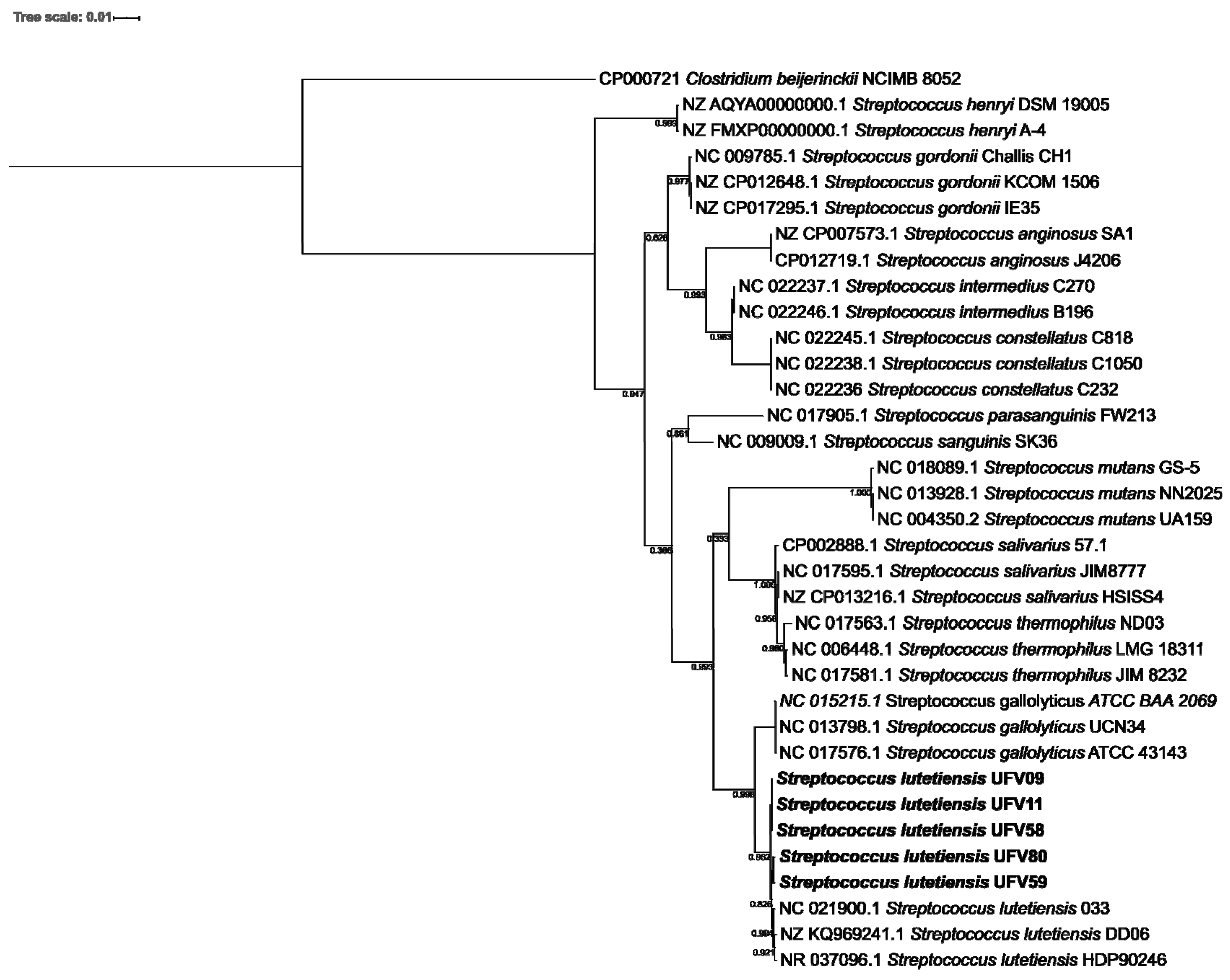
3.2. Streptococci Phylogeny
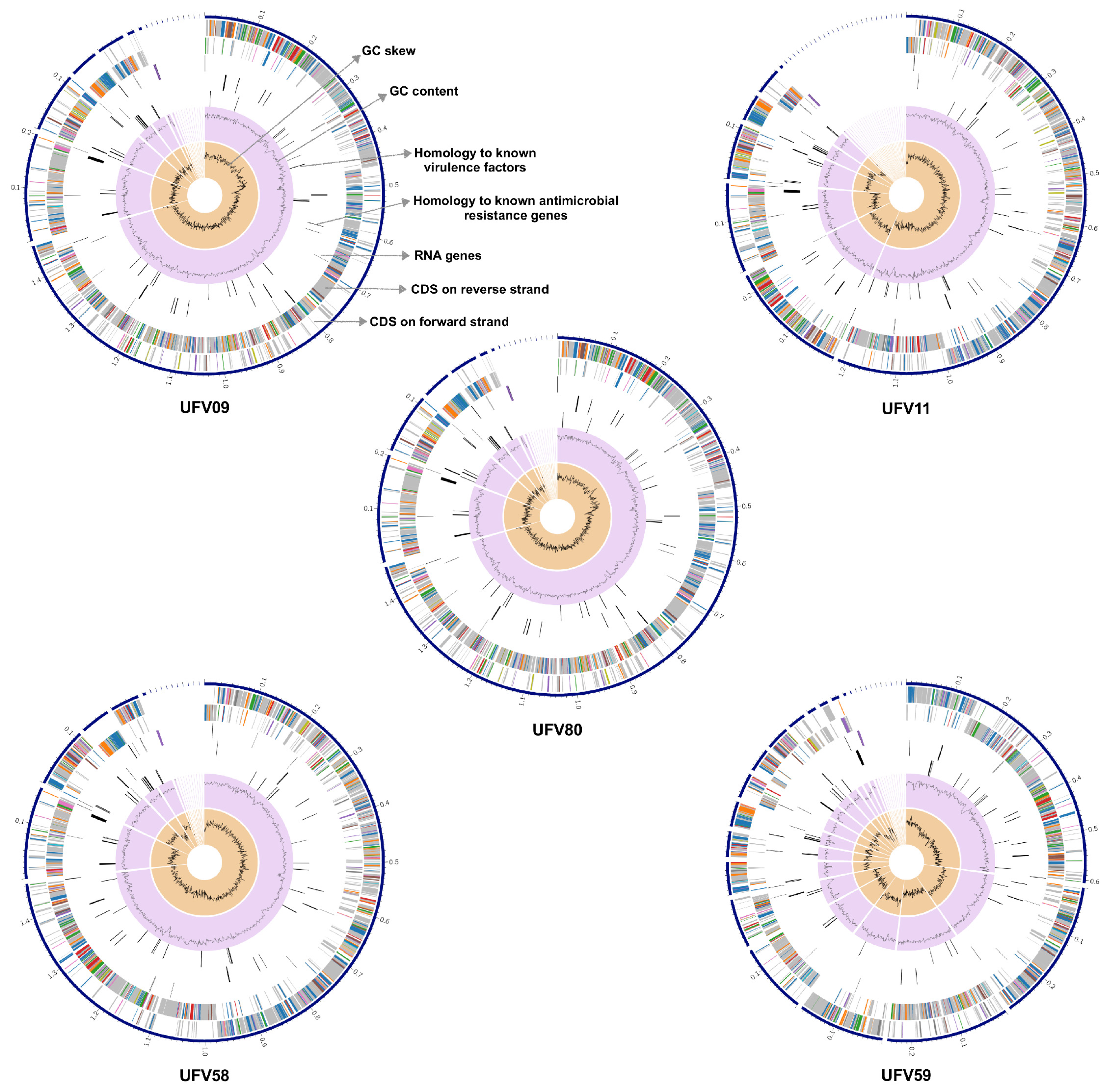
3.3. General Features of the Ruminal S. lutetiensis Genomes
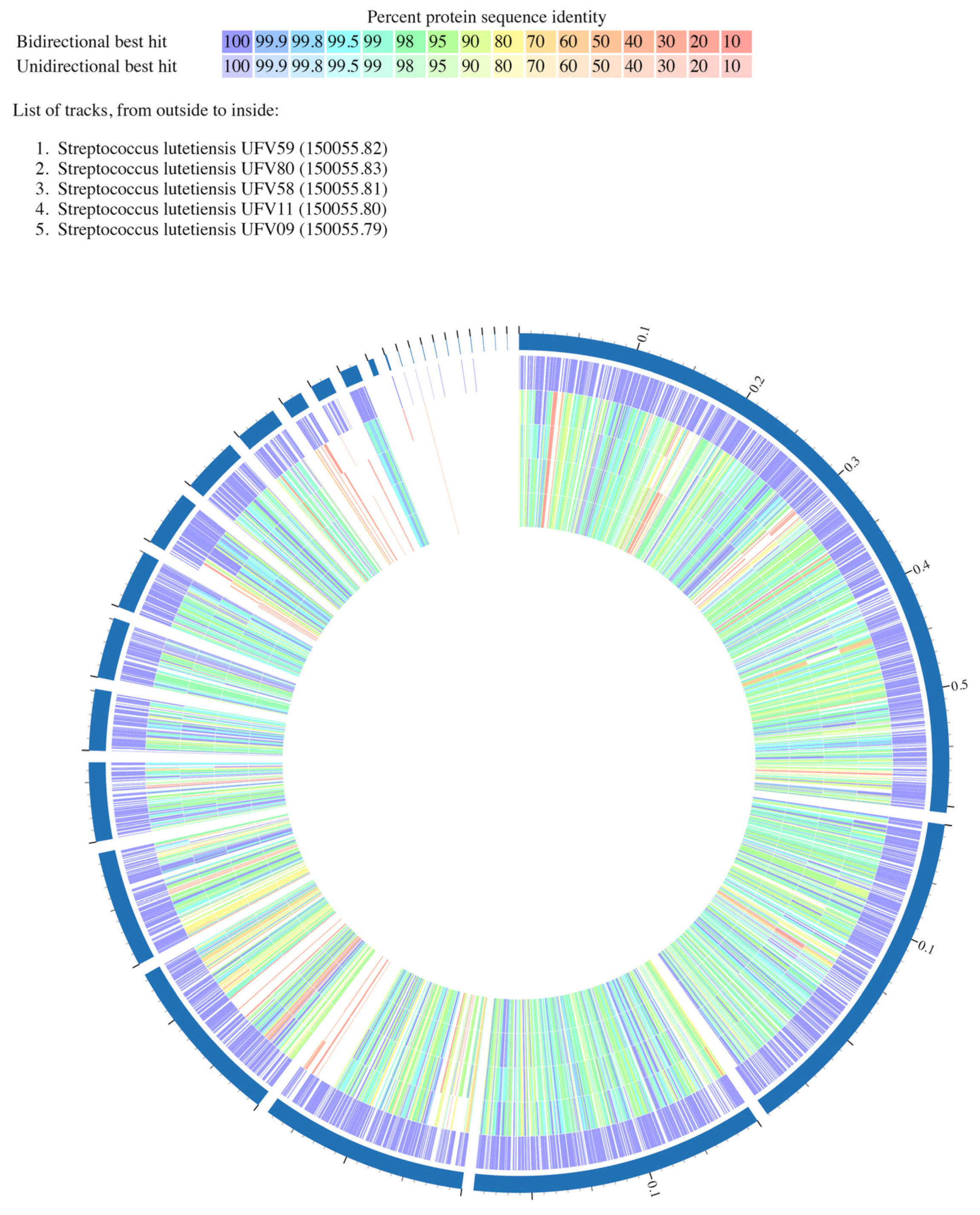
3.4. Analysis of the Predicted Proteomes from Ruminal S. lutetiensis
3.5. Functional Analysis of the Streptococcus lutetiensis Genomes
3.6. Analysis of CRISPR/Cas9 Systems in the Genomes of Ruminal S. lutetiensis
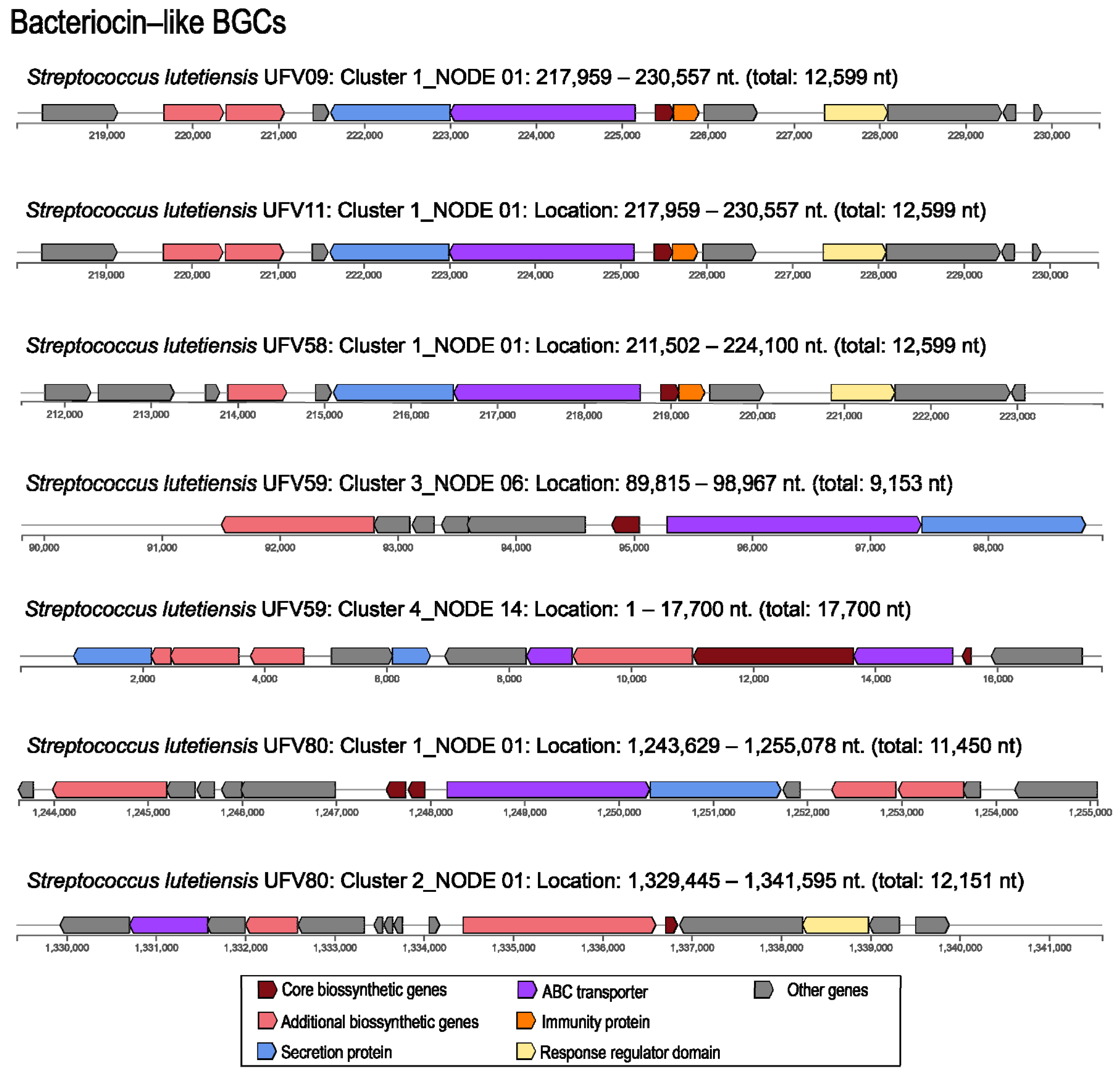
3.7. Genome Mining of Biosynthetic Gene Clusters for Bioactive Compounds
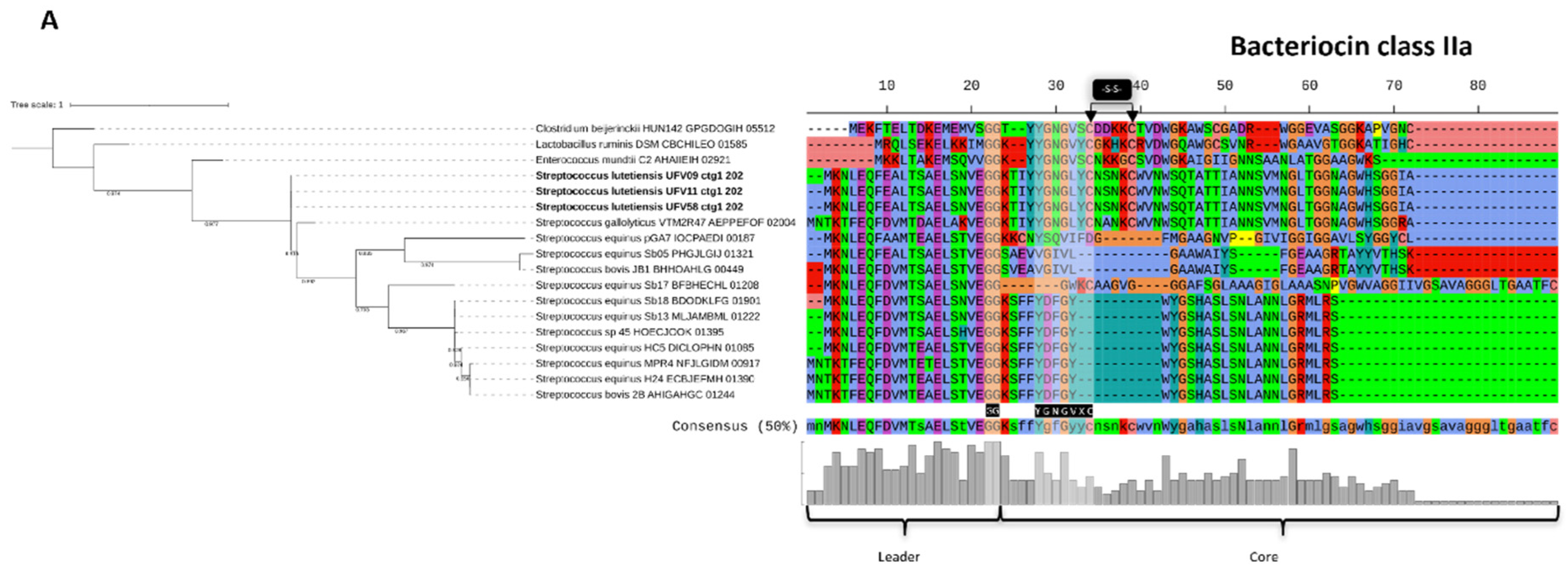
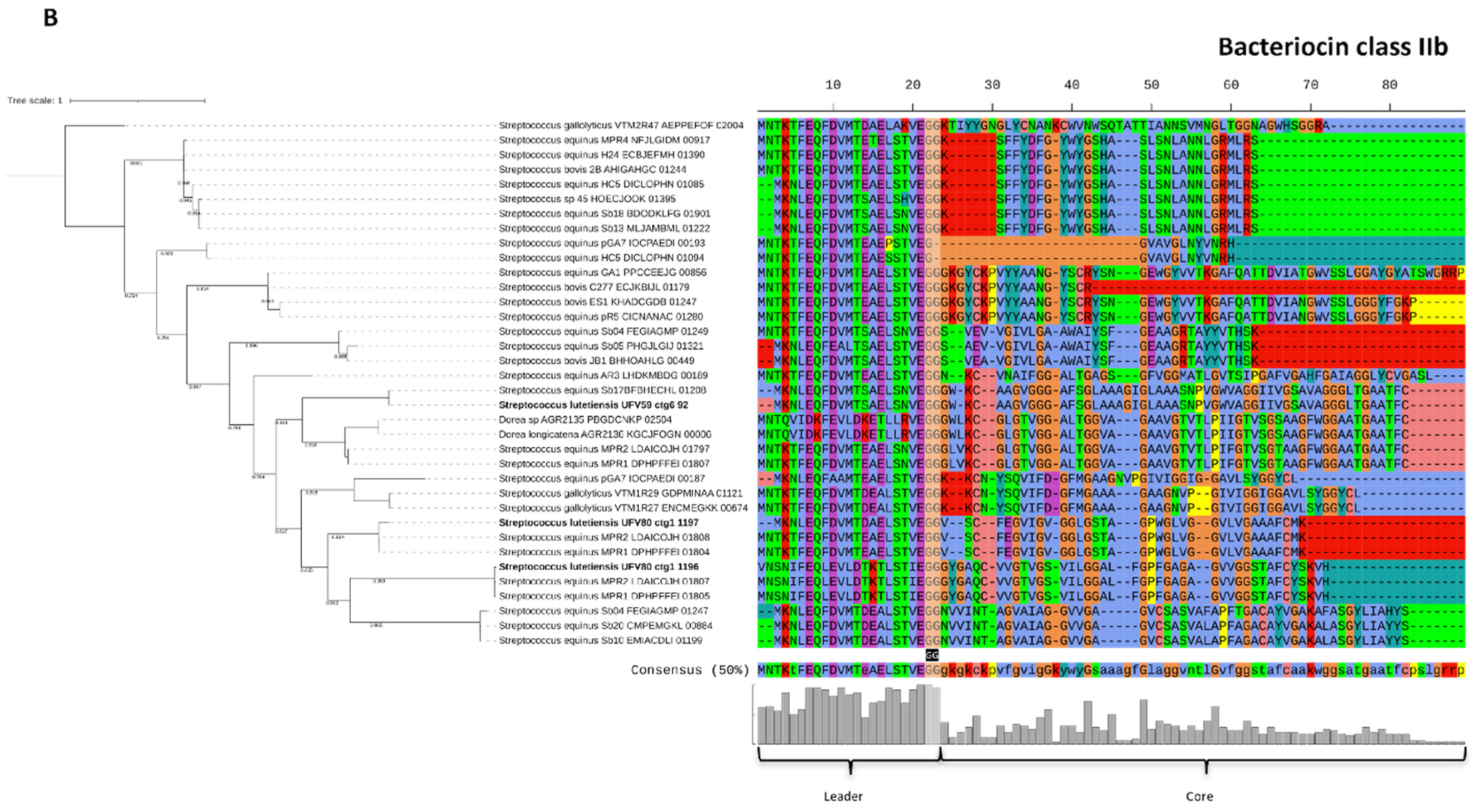
3.8. Characterization of the Bacteriocins Produced by Ruminal S. lutetiensis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matthews, C.; Crispie, F.; Lewis, E.; Reid, M.; O’Toole, P.W.; Cotter, P.D. The rumen microbiome: A crucial consideration when optimising milk and meat production and nitrogen utilisation efficiency. Gut Microbes 2019, 10, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Creevey, C.J.; Kelly, W.J.; Henderson, G.; Leahy, S.C. Determining the culturability of the rumen bacterial microbiome. Microb. Biotechnol. 2014, 7, 467–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letzel, A.-C.; Pidot, S.J.; Hertweck, C. Genome mining for ribosomally synthesized and post-translationally modified peptides (RiPPs) in anaerobic bacteria. BMC Genom. 2014, 15, 983. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, G.O.; Gruninger, R.J.; Badhan, A.; McAllister, T.A. Mining the rumen for fibrolytic feed enzymes. Anim. Front. 2016, 6, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Oyama, L.B.; Girdwood, S.E.; Cookson, A.R.; Fernandez-Fuentes, N.; Privé, F.; Vallin, H.E.; Wilkinson, T.J.; Golyshin, P.N.; Golyshina, O.V.; Mikut, R.; et al. The rumen microbiome: An underexplored resource for novel antimicrobial discovery. npj Biofilms Microbiomes 2017, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, S.M.; De Oliveira Mendes, T.A.; Santanta, M.F.; Huws, S.A.; Creevey, C.J.; Mantovani, H.C. Genomic and gene expression evidence of nonribosomal peptide and polyketide production among ruminal bacteria: A potential role in niche colonization? FEMS Microbiol. Ecol. 2019, 96, fiz198. [Google Scholar] [CrossRef] [PubMed]
- Odenyo, A.; Mackie, R.; Stahl, D.; White, B. The use of 16S rRNA-targeted oligonucleotide probes to study competition between ruminal fibrolytic bacteria: Development of probes for Ruminococcus species and evidence for bacteriocin production. Appl. Environ. Microbiol. 1994, 60, 3688–3696. [Google Scholar] [CrossRef] [Green Version]
- Rychlik, J.L.; Russell, J.B. Bacteriocin-like activity of Butyrivibrio fibrisolvens JL5 and its effect on other ruminal bacteria and ammonia production. Appl. Environ. Microbiol. 2002, 68, 1040–1046. [Google Scholar] [CrossRef] [Green Version]
- Nigutova, K.; Morovsky, M.; Pristas, P.; Teather, R.; Holo, H.; Javorsky, P. Production of enterolysin A by rumen Enterococcus faecalis strain and occurrence of enlA homologues among ruminal Gram-positive cocci. J. Appl. Microbiol. 2007, 102, 563–569. [Google Scholar] [CrossRef]
- Whitford, M.; McPherson, M.; Forster, R.; Teather, R. Identification of bacteriocin-like inhibitors from rumen Streptococcus spp. and isolation and characterization of bovicin 255. Appl. Environ. Microbiol. 2001, 67, 569–574. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, H.C.; Russell, J.B. Nisin resistance of Streptococcus bovis. Appl. Environ. Microbiol. 2001, 67, 808–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, A.C.; Bento, C.B.; Ruiz, J.C.; Queiroz, M.V.; Mantovani, H.C. Distribution and genetic diversity of bacteriocin gene clusters in rumen microbial genomes. Appl. Environ. Microbiol. 2015, 81, 7290–7304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Sieiro, P.; Montalbán-López, M.; Mu, D.; Kuipers, O.P. Bacteriocins of lactic acid bacteria: Extending the family. Appl. Microbiol. Biotechnol. 2016, 100, 2939–2951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, J.B.; Mantovani, H.C. The bacteriocins of ruminal bacteria and their potential as an alternative to antibiotics. J. Mol. Microbiol. Biotechnol. 2002, 4, 347–355. [Google Scholar]
- Lima, J.R.; Ribon, A.D.O.B.; Russell, J.B.; Mantovani, H.C. Bovicin HC5 inhibits wasteful amino acid degradation by mixed ruminal bacteria in vitro. FEMS Microbiol. Lett. 2009, 292, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Cotta, M.A.; Russell, J.B. Effect of peptides and amino acids on efficiency of rumen bacterial protein synthesis in continuous culture. J. Dairy Sci. 1982, 65, 226–234. [Google Scholar] [CrossRef]
- Macpherson, I. Soft agar techniques. In Tissue Culture; Elsevier: Amsterdam, The Netherlands, 1973; pp. 276–280. [Google Scholar]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Hamidian, M.; Hall, R.M. Acinetobacter baumannii ATCC 19606 carries GIsul2 in a genomic island located in the chromosome. Antimicrob. Agents Chemother. 2017, 61, e01991-16. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Woodbury, NY, USA, 1989. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Global Rumen Census, C.; Janssen, P.H. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project: Exploring the microbial part of ourselves in a changing world. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Ye, Y. Not all predicted CRISPR–Cas systems are equal: Isolated cas genes and classes of CRISPR like elements. BMC Bioinform. 2017, 18, 92. [Google Scholar] [CrossRef] [Green Version]
- Raden, M.; Ali, S.M.; Alkhnbashi, O.S.; Busch, A.; Costa, F.; Davis, J.A.; Eggenhofer, F.; Gelhausen, R.; Georg, J.; Heyne, S.; et al. Freiburg RNA tools: A central online resource for RNA-focused research and teaching. Nucleic Acids Res. 2018, 46, W25–W29. [Google Scholar] [CrossRef]
- Biswas, A.; Gagnon, J.N.; Brouns, S.J.; Fineran, P.C.; Brown, C.M. CRISPRTarget: Bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013, 10, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leplae, R.; Lima-Mendez, G.; Toussaint, A. ACLAME: A CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res. 2010, 38 (Suppl. 1), D57–D61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seshadri, R.; Leahy, S.C.; Attwood, G.T.; Teh, K.H.; Lambie, S.C.; Cookson, A.L.; Cookson, A.L.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Hadjithomas, M.; et al. Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat. Biotechnol. 2018, 36, 359. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Belguesmia, Y.; Naghmouchi, K.; Chihib, N.-E.; Drider, D. Class IIa Bacteriocins: Current Knowledge and Perspectives. In Prokaryotic Antimicrobial Peptides: From Genes to Applications; Drider, D., Rebuffat, S., Eds.; Springer: New York, NY, USA, 2011; pp. 171–195. [Google Scholar]
- Dirix, G.; Monsieurs, P.; Dombrecht, B.; Daniels, R.; Marchal, K.; Vanderleyden, J.; Michiels, J. Peptide signal molecules and bacteriocins in Gram-negative bacteria: A genome-wide in silico screening for peptides containing a double-glycine leader sequence and their cognate transporters. Peptides 2004, 25, 1425–1440. [Google Scholar] [CrossRef]
- Klaenhammer, T.R. Genetics of bacteriocins produced by lactic acid bacteria. FEMS Microbiol. Rev. 1993, 12, 39–85. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; Van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schagger, H. Tricine-SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, H.C.; Russell, J.B. The ability of a bacteriocin of Streptococcus bovis HC5 (bovicin HC5) to inhibit Clostridium aminophilum, an obligate amino acid fermenting bacterium from the rumen. Anaerobe 2002, 8, 247–252. [Google Scholar] [CrossRef]
- Pompilio, A.; Di Bonaventura, G.; Gherardi, G. An overview on Streptococcus bovis/Streptococcus equinus complex isolates: Identification to the species/subspecies level and antibiotic resistance. Int. J. Mol. Sci. 2019, 20, 480. [Google Scholar] [CrossRef] [Green Version]
- Jans, C.; Meile, L.; Lacroix, C.; Stevens, M.J. Genomics, evolution, and molecular epidemiology of the Streptococcus bovis/Streptococcus equinus complex (SBSEC). Infect. Genet. Evol. 2015, 33, 419–436. [Google Scholar] [CrossRef]
- Jans, C.; Boleij, A. The road to infection: Host-microbe interactions defining the pathogenicity of Streptococcus bovis/Streptococcus equinus complex members. Front. Microbiol. 2018, 9, 603. [Google Scholar] [CrossRef] [Green Version]
- Hill, D.; O’Connor, P.M.; Altermann, E.; Day, L.; Hill, C.; Stanton, C.; Ross, R.P. Extensive bacteriocin gene shuffling in the Streptococcus bovis/Streptococcus equinus complex reveals gallocin D with activity against vancomycin resistant enterococci. Sci. Rep. 2020, 10, 13431. [Google Scholar] [CrossRef]
- Heng, N.C.K.; Burtenshaw, G.A.; Jack, R.W.; Tagg, J.R.; Ubericin, A. A Class IIa Bacteriocin Produced by Streptococcus uberis. Appl. Environ. Microbiol. 2007, 73, 7763–7766. [Google Scholar] [CrossRef] [Green Version]
- Nes, I.F.; Holo, H. Class II antimicrobial peptides from lactic acid bacteria. Pept. Sci. 2000, 55, 50–61. [Google Scholar] [CrossRef]
- Zimina, M.; Babich, O.; Prosekov, A.; Sukhikh, S.; Ivanova, S.; Shevchenko, M.; Noskova, S. Overview of global trends in classification, methods of preparation and application of bacteriocins. Antibiotics 2020, 9, 553. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, R.E.; Klesse, N.A.; Jack, R.W.; Tagg, J.R. Molecular and genetic characterization of a novel nisin variant produced by Streptococcus uberis. Appl. Environ. Microbiol. 2006, 72, 1148–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyink, O.; Balakrishnan, M.; Tagg, J.R. Streptococcus rattus strain BHT produces both a class I two-component lantibiotic and a class II bacteriocin. FEMS Microbiol. Lett. 2005, 252, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadimitriou, K.; Anastasiou, R.; Mavrogonatou, E.; Blom, J.; Papandreou, N.C.; Hamodrakas, S.J.; Ferreira, S.; Renault, P.; Supply, P.; Pot, B.; et al. Comparative genomics of the dairy isolate Streptococcus macedonicus ACA-DC 198 against related members of the Streptococcus bovis/Streptococcus equinus complex. BMC Genom. 2014, 15, 272. [Google Scholar] [CrossRef] [Green Version]
- Vuppada, R.K.; Hansen, C.R.; Strickland, K.A.; Kelly, K.M.; McCleary, W.R. Phosphate signaling through alternate conformations of the PstSCAB phosphate transporter. BMC Microbiol. 2018, 18, 8. [Google Scholar] [CrossRef]
- Carmany, D.O.; Hollingsworth, K.; McCleary, W.R. Genetic and biochemical studies of phosphatase activity of PhoR. J. Bacteriol. 2003, 185, 1112–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, R.J. Ruminal microbiology, biotechnology, and ruminant nutrition: Progress and problems. J. Anim. Sci. 1994, 72, 2992–3003. [Google Scholar] [CrossRef] [Green Version]
- Le Rhun, A.; Escalera-Maurer, A.; Bratovič, M.; Charpentier, E. CRISPR-Cas in Streptococcus pyogenes. RNA Biol. 2019, 16, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Chylinski, K.; Makarova, K.S.; Charpentier, E.; Koonin, E.V. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 2014, 42, 6091–6105. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | ||||||||
|---|---|---|---|---|---|---|---|---|
| Isolates Identification (IDs) | Citrobacter freudii ATCC 8090 | Enterococcus faecalis ATCC 4083 | Escherichia coli ATCC 10536 | Lactobacillus paracasei subsp. paracasei ATCC 355 | Listeria monocytogenes ATCC 7644 | Proteus vulgaris ATCC 13315 | Salmonella enterica serovar Typhimurium ATCC 14028 | Staphylococcus aureus ATCC 29213 |
| UFV9 | - | - | ++ | - | + | - | ++ | ++ |
| UFV11 | - | - | ++ | - | +++ | - | ++ | + |
| UFV58 | - | - | ++ | + | +++ | - | ++ | - |
| UFV59 | - | ++ | ++ | - | - | - | +++ | - |
| UFV80 | - | + | ++ | + | - | - | +++ | ++ |
| UFV09 | UFV11 | UFV58 | UFV59 | UFV80 | ||
|---|---|---|---|---|---|---|
| Species (rRNA 16S gene sequences) | Streptococcus lutetiensis | Streptococcus lutetiensis | Streptococcus lutetiensis | Streptococcus lutetiensis | Streptococcus lutetiensis | |
| Source | Nellore rumen | Nellore rumen | Nellore rumen | Nellore rumen | Nellore rumen | |
| Genome assembly | N of reads | 261881 | 170826 | 219215 | 158064 | 166894 |
| Contigs | 21 | 33 | 16 | 28 | 18 | |
| L50 | 1 | 1 | 1 | 3 | 1 | |
| N50 | 1451917 | 1214823 | 1463551 | 248939 | 1460340 | |
| Genome Size (bp) | 1849765 | 1854024 | 1854488 | 1977774 | 1902765 | |
| GC Content (%) | 38.00 | 38.03 | 38.05 | 37.43 | 37.88 | |
| Plasmids | - | - | - | - | - | |
| Chromosomes | 1 | 1 | 1 | 1 | 1 | |
| Genome annotation | PATRIC ID | 150055.79 | 150055.80 | 150055.81 | 150055.82 | 150055.83 |
| CDS | 1793 | 1808 | 1824 | 1903 | 1860 | |
| tRNA | 50 | 50 | 51 | 48 | 49 | |
| rRNA | 5 | 5 | 5 | 5 | 6 | |
| crispr repeat | - | - | - | - | 29 | |
| crispr spacer | - | - | - | - | 28 | |
| crispr array | - | - | - | - | 1 | |
| Protein features | Hypothetical proteins | 296 (16.51%) | 299 (16.54%) | 322 (17.65%) | 362 (19.02%) | 312 (16.77%) |
| Proteins with functional assignments | 1497 (83.49%) | 1509 (83.46%) | 1502 (82.35%) | 1541 (80.98%) | 1548 (83.23%) | |
| Proteins with EC number assignments | 0 | 0 | 0 | 0 | 0 | |
| Proteins with GO assignments | 473 | 481 | 474 | 481 | 477 | |
| Proteins with Pathway assignments | 0 | 0 | 0 | 0 | 0 | |
| Proteins with Subsystem assignments | 0 | 0 | 0 | 0 | 0 | |
| Proteins with PATRIC genus-specific family (PLfam) assignments | 1748 | 1754 | 1766 | 1858 | 1779 | |
| Proteins with PATRIC cross-genus family (PGfam) assignments | 1756 | 1767 | 1771 | 1863 | 1787 | |
| Proteins with FIGfam assignments | 0 | 0 | 0 | 0 | 0 | |
| Specialty genes | Antibiotic Resistance (PATRIC) | 22 | 22 | 22 | 22 | 23 |
| Drug Target (DrugBank) | 10 | 14 | 10 | 10 | 9 | |
| Drug Target (TTD) | 1 | 1 | 2 | 1 | 1 | |
| Transporter (TCDB) | 16 | 16 | 15 | 17 | 14 | |
| Virulence Factor (PATRIC VF) | - | 2 | 1 | 1 | - | |
| Virulence Factor (VFDB) | 5 | 6 | 4 | 5 | 5 | |
| Virulence Factor (Victors) | 29 | 32 | 31 | 32 | 31 | |
| Bacteriocin cluster (antiSMASH) | 1 | 1 | 1 | 2 | 2 | |
| NRPS (antiSMASH) | 1 | 1 | 1 | 1 | - | |
| T3PKS (antiSMASH) | 1 | 1 | 1 | 1 | 1 | |
| Furan (antiSMASH) | 1 | 1 | ||||
| Arylpolyene (antiSMASH) | 1 | 1 | 1 | 1 | 1 | |
| ENA ID | Genome accession n° | ERR5159231 | ERR5159232 | ERR5159233 | ERR5159234 | ERR5159235 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira, I.M.F.; Godoy-Santos, F.; Oyama, L.B.; Moreira, S.M.; Dias, R.G.; Huws, S.A.; Creevey, C.J.; Mantovani, H.C. Whole-Genome Sequencing and Comparative Genomic Analysis of Antimicrobial Producing Streptococcus lutetiensis from the Rumen. Microorganisms 2022, 10, 551. https://doi.org/10.3390/microorganisms10030551
de Oliveira IMF, Godoy-Santos F, Oyama LB, Moreira SM, Dias RG, Huws SA, Creevey CJ, Mantovani HC. Whole-Genome Sequencing and Comparative Genomic Analysis of Antimicrobial Producing Streptococcus lutetiensis from the Rumen. Microorganisms. 2022; 10(3):551. https://doi.org/10.3390/microorganisms10030551
Chicago/Turabian Stylede Oliveira, Isabela Maria Fernandes, Fernanda Godoy-Santos, Linda Boniface Oyama, Sofia Magalhães Moreira, Rodrigo Gonçalves Dias, Sharon Ann Huws, Christopher J. Creevey, and Hilário Cuquetto Mantovani. 2022. "Whole-Genome Sequencing and Comparative Genomic Analysis of Antimicrobial Producing Streptococcus lutetiensis from the Rumen" Microorganisms 10, no. 3: 551. https://doi.org/10.3390/microorganisms10030551
APA Stylede Oliveira, I. M. F., Godoy-Santos, F., Oyama, L. B., Moreira, S. M., Dias, R. G., Huws, S. A., Creevey, C. J., & Mantovani, H. C. (2022). Whole-Genome Sequencing and Comparative Genomic Analysis of Antimicrobial Producing Streptococcus lutetiensis from the Rumen. Microorganisms, 10(3), 551. https://doi.org/10.3390/microorganisms10030551