
Long-Read Sequencing and Hybrid Assembly for Genomic Analysis of Clinical Brucella melitensis Isolates

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolate Collection, DNA Extraction, and Sequencing
2.2. Short Reads Assembly
2.3. Long Reads Assembly
2.4. Hybrid Assembly
2.5. Downstream Analyses
3. Results
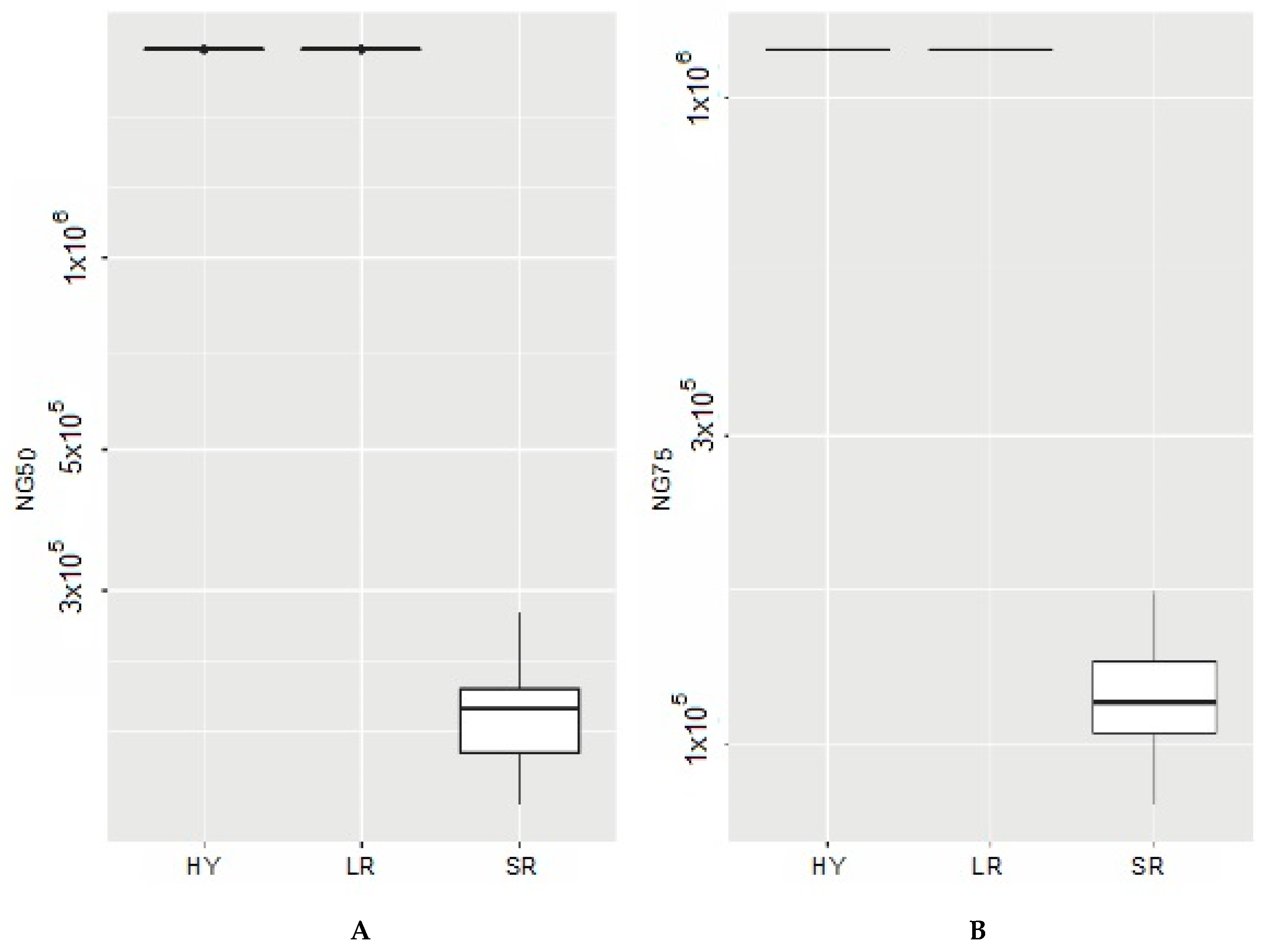
3.1. Read and Assembly Statistics
3.2. Virulence Gene Identification
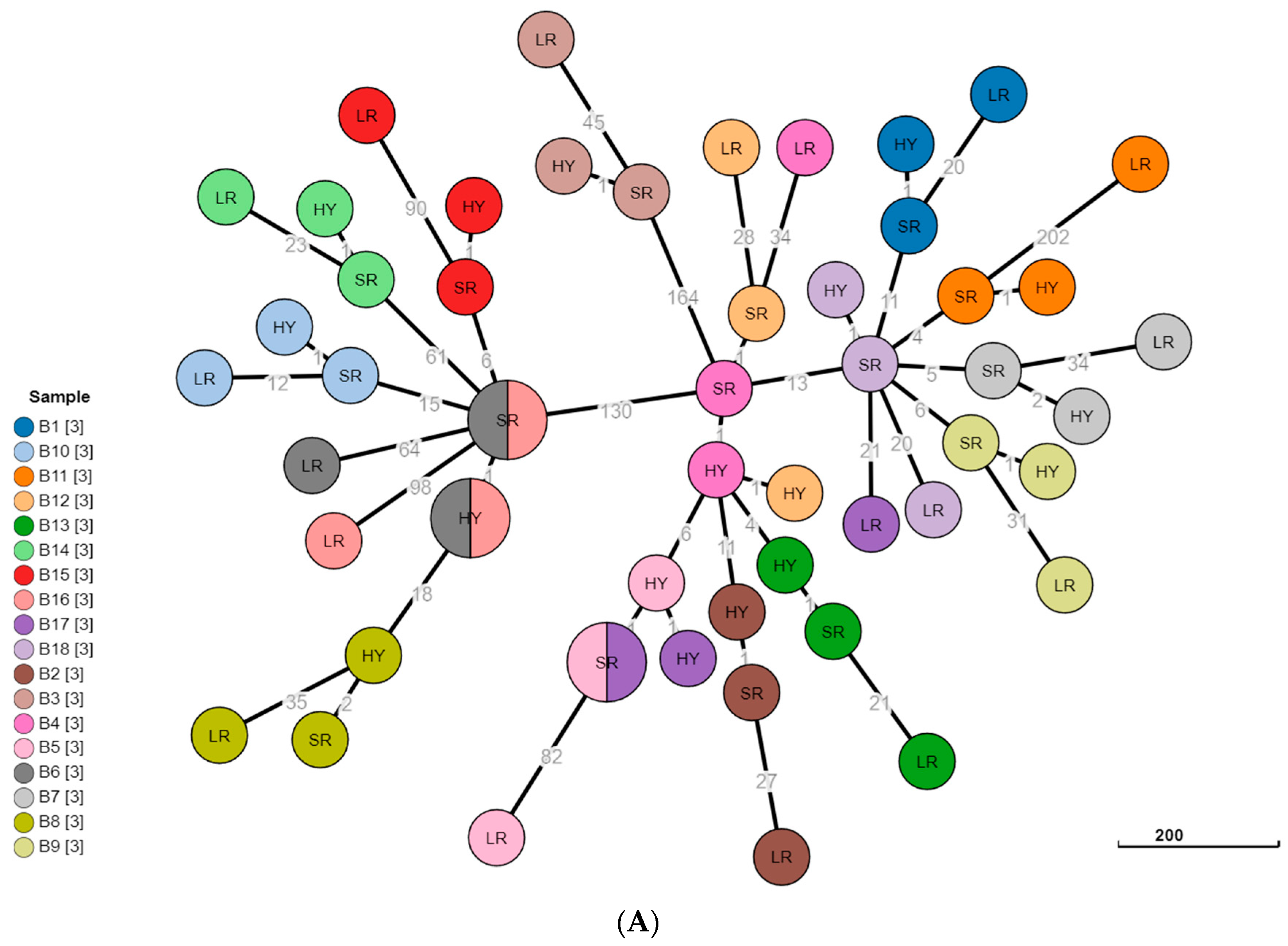
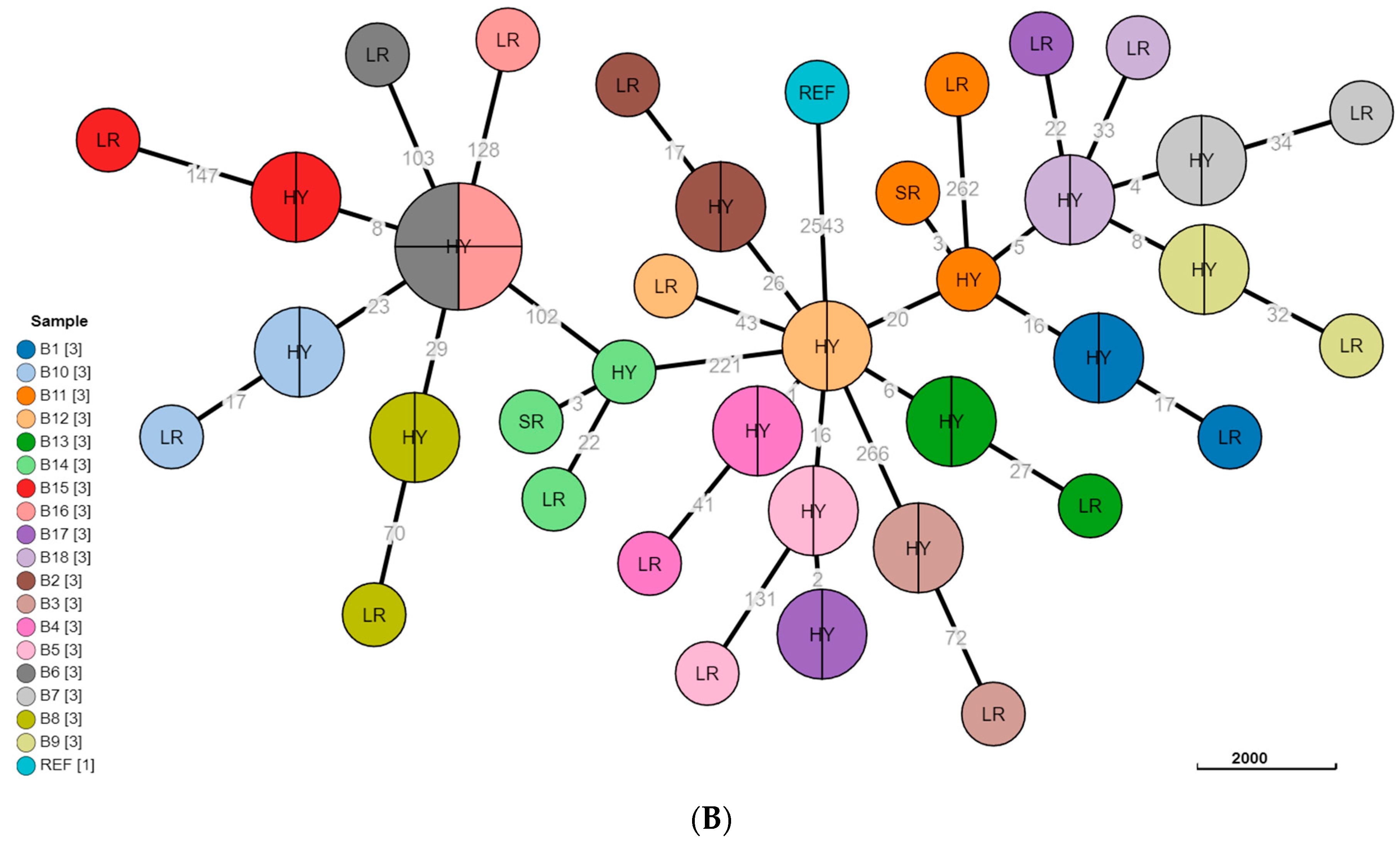
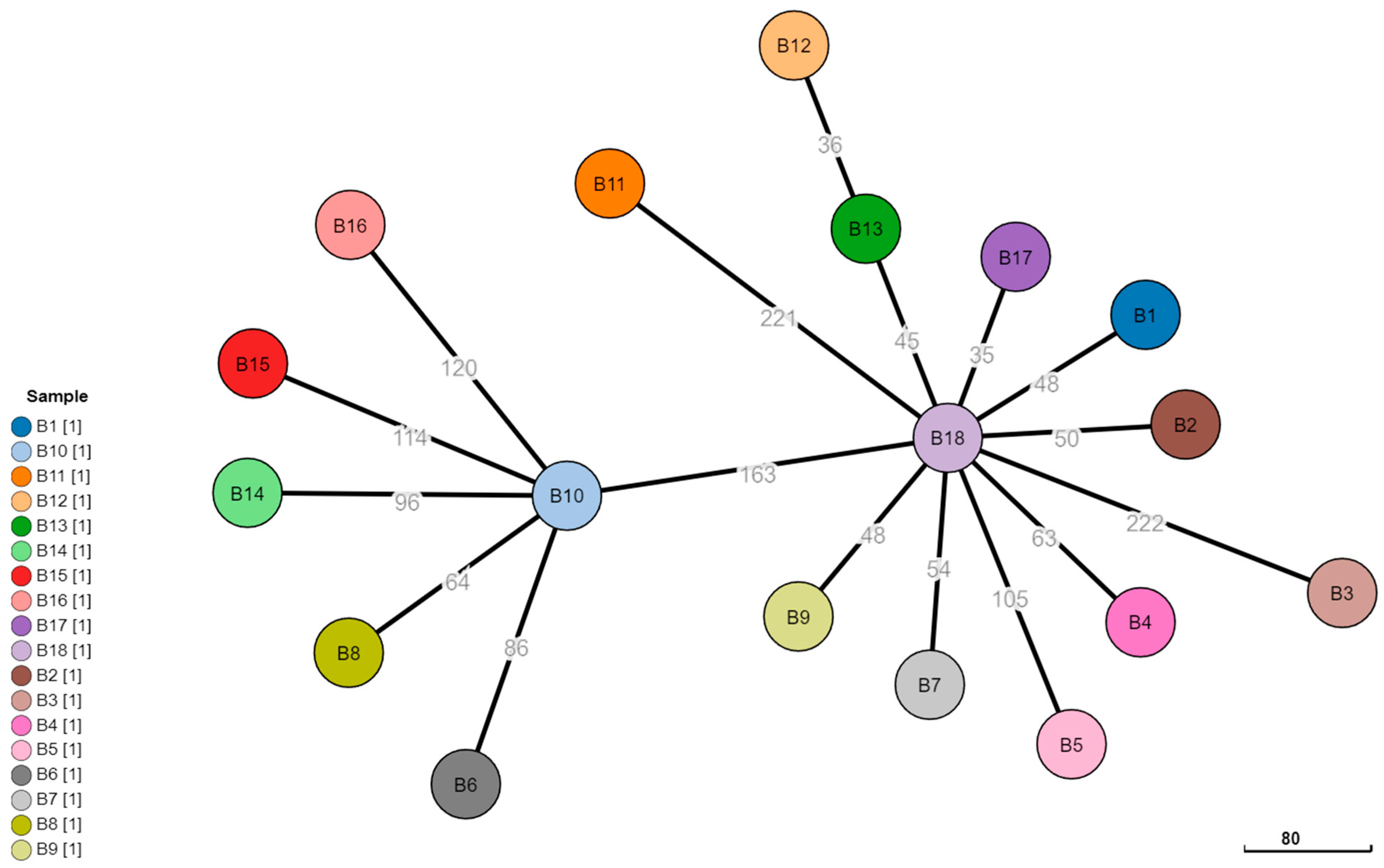
3.3. Phylogeny Comparison
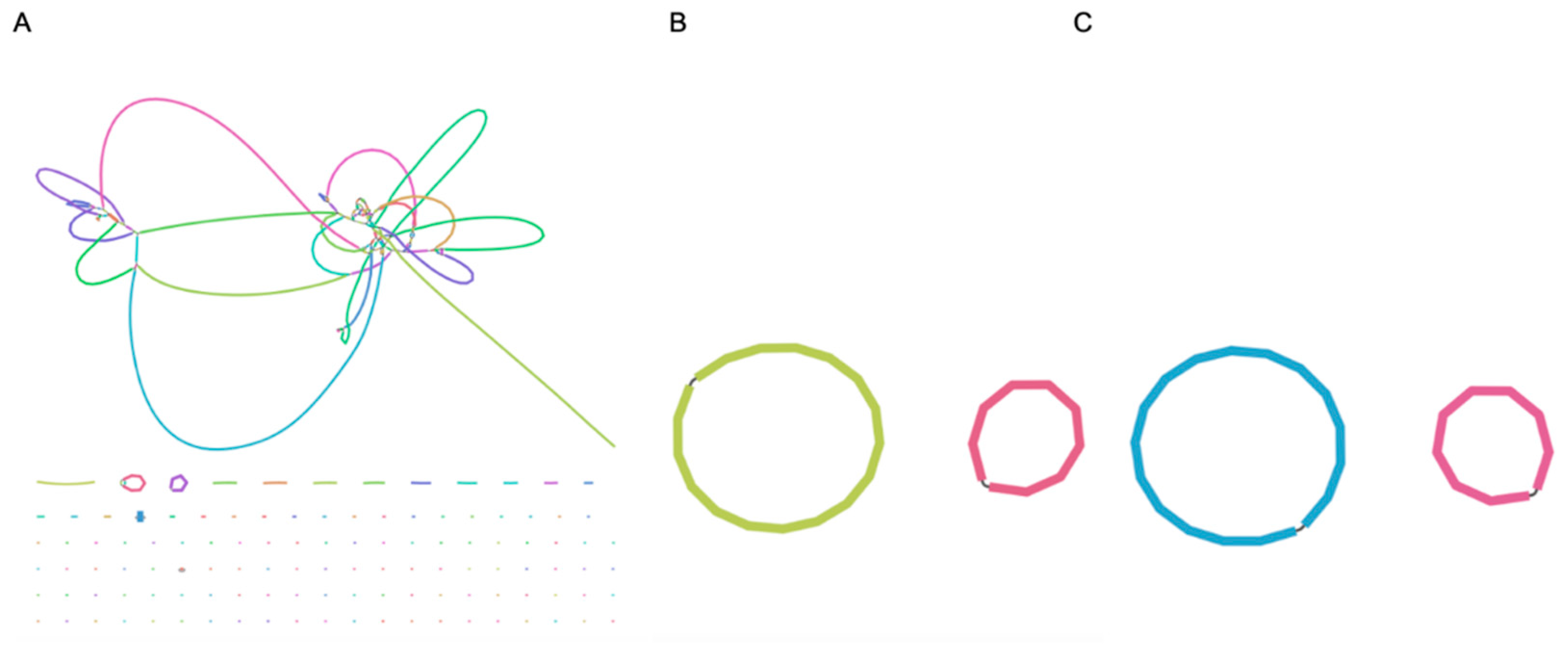
3.4. What Is in the Gaps?
4. Discussion
4.1. Study Summary
4.2. Implications for Field-Deployed or Low-Resource Settings
4.3. Limitations and Future Research
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bachmann, N.L.; Rockett, R.J.; Timms, V.J.; Sintchenko, V. Advances in clinical sample preparation for identification and characterization of bacterial pathogens using metagenomics. Front. Public Health 2018, 6, 363. [Google Scholar] [CrossRef] [PubMed]
- Gardy, J.L.; Loman, N.J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef] [PubMed]
- De Maio, N.; Shaw, L.P.; Hubbard, A.; George, S.; Sanderson, N.D.; Swann, J.; Wick, R.; AbuOun, M.; Stubberfield, E.; Hoosdally, S.J.; et al. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microb. Genom. 2019, 5, e000294. [Google Scholar] [CrossRef] [PubMed]
- Magi, A.; Semeraro, R.; Mingrino, A.; Giusti, B.; D’Aurizio, R. Nanopore sequencing data analysis: State of the art, applications and challenges. Brief. Bioinform. 2018, 19, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Tyler, A.D.; Mataseje, L.; Urfano, C.J.; Schmidt, L.; Antonation, K.S.; Mulvey, M.R.; Corbett, C.R. Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Sci. Rep. 2018, 8, 10931. [Google Scholar] [CrossRef] [Green Version]
- Susilawati, T.N.; Jex, A.R.; Cantacessi, C.; Pearson, M.; Navarro, S.; Susianto, A.; Loukas, A.C.; McBride, W.J.H. Deep sequencing approach for investigating infectious agents causing fever. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Jiang, W.; Shi, Y.; Ye, H.; Lin, J. Applications of sequencing technology in clinical microbial infection. J. Cell Mol. Med. 2019, 23, 7143–7150. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Kuang, D.; Xu, X.; González-Escalona, N.; Erickson, D.L.; Brown, E.; Meng, J. Genomic Analyses of Multidrug-Resistant Salmonella Indiana, Typhimurium, and Enteritidis Isolates Using MinION and MiSeq Sequencing Technologies. PLoS ONE 2020, 15, e0235641. [Google Scholar] [CrossRef]
- Alghoribi, M.F.; Zidan, K.H.; Alswaji, A.A.; Alhafufi, A.N.; Ahmed, A.; Balkhy, H.H. Whole-Genome Sequence of a Brucella melitensis Strain Isolated from Sheep in Saudi Arabia. Microbiol. Resour. Announc. 2018, 7, e01189-18. [Google Scholar] [CrossRef] [Green Version]
- Aljanazreh, B.; Alzatari, K.; Tamimi, A.; Alsaafeen, M.H.; Hassouneh, W.; Ashhab, Y. Brucellosis re-emergence after a decade of quiescence in Palestine, 2015–2017: A seroprevalence and molecular characterization study. Transbound. Emerg. Dis. 2021. [Google Scholar] [CrossRef]
- Anis, E.; Leventhal, A.; Grotto, I.; Gandacu, D.; Warshavsky, B.; Shimshony, A.; Israeli, A. Recent trends in human brucellosis in Israel. Isr. Med Assoc. J. IMAJ 2011, 13, 359–365. [Google Scholar] [PubMed]
- Rabinowitz, P.; Zilberman, B.; Motro, Y.; Roberts, M.C.; Greninger, A.; Nesher, L.; Ben-Shimol, S.; Yagel, Y.; Gdalevich, M.; Sagi, O.; et al. Whole Genome Sequence Analysis of Brucella melitensis Phylogeny and Virulence Factors. Microbiol. Res. 2021, 12, 698–710. [Google Scholar] [CrossRef]
- Bardenstein, S.; Gibbs, R.E.; Yagel, Y.; Motro, Y.; Moran-Gilad, J. Brucellosis Outbreak Traced to Commercially Sold Camel Milk through Whole-Genome Sequencing, Israel. Emerg. Infect. Dis. 2021, 27, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, B.; Motro, Y.; Sagi, O.; Kornspan, D.; Ben-Shimol, S.; Gdalevich, M.; Yagel, Y.; Davidovitch, N.; Khalfin, B.; Rabinowitz, P.; et al. Genomic Epidemiology of Clinical Brucella Melitensis Isolates from Southern Israel. Microorganisms 2022, 10, 238. [Google Scholar] [CrossRef]
- Vered, O.; Simon-Tuval, T.; Yagupsky, P.; Malul, M.; Cicurel, A.; Davidovitch, N. The Price of a Neglected Zoonosis: Case-Control Study to Estimate Healthcare Utilization Costs of Human Brucellosis. PLoS ONE 2015, 10, e0145086. [Google Scholar] [CrossRef] [Green Version]
- Mongkolrattanothai, K.; Naccache, S.N.; Bender, J.M.; Samayoa, E.; Pham, E.; Yu, G.; Dien Bard, J.; Miller, S.; Aldrovandi, G.; Chiu, C.Y. Neurobrucellosis: Unexpected Answer From Metagenomic Next-Generation Sequencing. J. Pediatr. Infect. Dis. Soc. 2017, 6, 393–398. [Google Scholar] [CrossRef]
- Avijgan, M.; Rostamnezhad, M.; Jahanbani-Ardakani, H. Clinical and Serological Approach to Patients with Brucellosis: A Common Diagnostic Dilemma and a Worldwide Perspective. Microb. Pathog. 2019, 129, 125–130. [Google Scholar] [CrossRef]
- Mantur, B.G.; Amarnath, S.K.; Shinde, R.S. Review of Clinical and Laboratory Features of Human Brucellosis. Indian J. Med. Microbiol. 2007, 25, 188–202. [Google Scholar] [CrossRef]
- Al Dahouk, S.; Nöckler, K. Implications of Laboratory Diagnosis on Brucellosis Therapy. Expert Rev. Anti Infect. Ther. 2011, 9, 833–845. [Google Scholar] [CrossRef]
- Brunker, K.; Marston, D.A.; Horton, D.L.; Cleaveland, S.; Fooks, A.R.; Kazwala, R.; Ngeleja, C.; Lembo, T.; Sambo, M.; Mtema, Z.J.; et al. Elucidating the phylodynamics of endemic rabies virus in eastern Africa using whole-genome sequencing. Virus Evol. 2015, 1, vev011. [Google Scholar] [CrossRef] [Green Version]
- Greninger, A.L.; Naccache, S.N.; Federman, S.; Yu, G.; Mbala, P.; Bres, V.; Stryke, D.; Bouquet, J.; Somasekar, S.; Linnen, J.M.; et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 2015, 7, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolotin, V.; Kovalenko, G.; Marchenko, N.; Solodiankin, O.; Rudova, N.; Kutsenko, V.; Bortz, E.; Gerilovych, A.; Drown, D.M. Complete Genome Sequence of Brucella abortus 68, Isolated from Aborted Fetal Sheep in Ukraine. Microbiol. Resour. Announc. 2021, 10, e01436-20. [Google Scholar] [CrossRef] [PubMed]
- Gündoğdu, A.; Ulu-Kilic, A.; Kilic, H.; Nalbantoglu, O.U. Rapid detection of difficult-to-culture bacterial pathogens using real-time nanopore sequencing. Infect. Dis. Clin. Microbiol. 2019, 1, 128–133. [Google Scholar] [CrossRef] [Green Version]
- Babraham Bioinformatics. FastQC. A Quality Control tool for High Throughput Sequence Data. 2021. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 29 December 2021).
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinforma. 2020, 70, e102. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- INNUca.py. Bioinformatics @ Molecular Microbiology and Infection Unit. 2021. Available online: https://github.com/B-UMMI/INNUca (accessed on 29 December 2021).
- Leger, A.; Leonardi, T. pycoQC, interactive quality control for Oxford Nanopore Sequencing. J. Open Source Softw. 2019, 4, 1236. [Google Scholar] [CrossRef]
- Wick, R. Porechop. 2021. Available online: https://github.com/rrwick/Porechop (accessed on 29 December 2021).
- Wick, R. rrwick/Filtlong. 2021. Available online: https://github.com/rrwick/Filtlong (accessed on 29 December 2021).
- Wick, R.R.; Holt, K.E. Benchmarking of long-read assemblers for prokaryote whole genome sequencing. F1000Research 2020, 8, 2138. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Cerdeira, L.T.; Hawkey, J.; Méric, G.; Vezina, B.; Wyres, K.L.; Holt, K.E. Trycycler: Consensus long-read assemblies for bacterial genomes. Genome Biol. 2021, 22, 266. [Google Scholar] [CrossRef]
- Wick, R. Trycycler. GitHub. Available online: https://github.com/rrwick/Trycycler (accessed on 29 December 2021).
- Wick, R.R. Trycycler. Zenodo. 2021. Available online: https://zenodo.org/record/5769082 (accessed on 29 December 2021).
- Petit, R.A., III. Dragonflye. 2022. Available online: https://github.com/rpetit3/dragonflye (accessed on 30 December 2021).
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Šikić, M. Time- and Memory-Efficient Genome Assembly with Raven. Nat. Comput. Sci. 2021, 1, 332–336. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies. Medaka. 2022. Available online: https://github.com/nanoporetech/medaka (accessed on 30 December 2021).
- Li, H. lh3/miniasm. 2021. Available online: https://github.com/lh3/miniasm (accessed on 29 December 2021).
- Wick, R.R.; Holt, K.E. Polypolish: Short-Read Polishing of Long-Read Bacterial Genome Assemblies. PLoS Comput. Biol. 2022, 18, e1009802. [Google Scholar] [CrossRef]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA Genome Assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. mlst. 2021. Available online: https://github.com/tseemann/mlst (accessed on 29 December 2021).
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simao, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. 22 June 2021. Available online: http://arxiv.org/abs/2106.11799 (accessed on 14 December 2021).
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Khelik, K.; Lagesen, K.; Sandve, G.K.; Rognes, T.; Nederbragt, A.J. NucDiff: In-depth characterization and annotation of differences between two sets of DNA sequences. BMC Bioinform. 2017, 18, 338. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [Green Version]
- Bradnam, K.R.; Fass, J.N.; Alexandrov, A.; Baranay, P.; Bechner, M.; Birol, I.; Boisvert, S.; Chapman, J.A.; Chapuis, G.; Chikhi, R.; et al. Assemblathon 2: Evaluating de novo methods of genome assembly in three vertebrate species. GigaScience 2013, 2, 10. [Google Scholar] [CrossRef]
- Silva, M.; Machado, M.P.; Silva, D.N.; Rossi, M.; Moran-Gilad, J.; Santos, S.; Ramirez, M.; Carrico, J.A. chewBBACA: A complete suite for gene-by-gene schema creation and strain identification. Microb. Genomics 2018, 4, e000166. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Alikhan, N.-F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carriço, J.A.; Achtman, M. GrapeTree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018, 28, 1395–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Snippy. 2022. Available online: https://github.com/tseemann/snippy (accessed on 30 December 2021).
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid Phylogenetic Analysis of Large Samples of Recombinant Bacterial Whole Genome Sequences Using Gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janowicz, A.; De Massis, F.; Ancora, M.; Cammà, C.; Patavino, C.; Battisti, A.; Prior, K.; Harmsen, D.; Scholz, H.; Zilli, K.; et al. Core Genome Multilocus Sequence Typing and Single Nucleotide Polymorphism Analysis in the Epidemiology of Brucella Melitensis Infections. J. Clin. Microbiol. 2018, 56, e00517-18. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Therrien, D.A.; Konganti, K.; Gill, J.J.; Davis, B.W.; Hillhouse, A.E.; Michalik, J.; Cross, H.R.; Smith, G.C.; Taylor, T.M.; Riggs, P.K. Complete Whole Genome Sequences of Escherichia coli Surrogate Strains and Comparison of Sequence Methods with Application to the Food Industry. Microorganisms 2021, 9, 608. [Google Scholar] [CrossRef]
- Chen, Z.; Erickson, D.L.; Meng, J. Polishing the Oxford Nanopore long-read assemblies of bacterial pathogens with Illumina short reads to improve genomic analyses. Genomics 2021, 113, 1366–1377. [Google Scholar] [CrossRef]
- Khezri, A.; Avershina, E.; Ahmad, R. Hybrid Assembly Provides Improved Resolution of Plasmids, Antimicrobial Resistance Genes, and Virulence Factors in Escherichia coli and Klebsiella pneumoniae Clinical Isolates. Microorganisms 2021, 9, 2560. [Google Scholar] [CrossRef]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.C.; Zwirglmaier, K.; Vette, P.; Holowachuk, S.A.; Stoecker, K.; Genzel, G.H.; Antwerpen, M.H. MinION as part of a biomedical rapidly deployable laboratory. J. Biotechnol. 2017, 250, 16–22. [Google Scholar] [CrossRef]
- Georgi, E.; Walter, M.C.; Pfalzgraf, M.-T.; Northoff, B.H.; Holdt, L.M.; Scholz, H.C.; Zoeller, L.; Zange, S.; Antwerpen, M.H. Whole genome sequencing of Brucella melitensis isolated from 57 patients in Germany reveals high diversity in strains from Middle East. PLoS ONE 2017, 12, e0175425. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrid Assemblies | ||||
| Mean | Median | Minimum | Maximum | |
| Total length (bp) | 3.31 × 106 | 3.31 × 106 | 3.31 × 106 | 3.31 × 106 |
| Largest contig (bp) | 2.13 × 106 | 2.13 × 106 | 2.13 × 106 | 2.13 × 106 |
| NG50 (bp) | 2.13 × 106 | 2.13 × 106 | 2.13 × 106 | 2.13 × 106 |
| NG75 (bp) | 1.19 × 106 | 1.19E × 106 | 1.19 × 106 | 1.19 × 106 |
| Average coverage depth (x) | 115.7 | 110.0 | 42.0 | 177.0 |
| BUSCO completeness (%) | 97.9 | 98.0 | 97.3 | 98.0 |
| Long-Read Assemblies | ||||
| Mean | Median | Minimum | Maximum | |
| Total length (bp) | 3.32 × 106 | 3.32 × 106 | 3.31 × 106 | 3.32 × 106 |
| Largest contig (bp) | 2.13 × 106 | 2.13 × 106 | 2.13 × 106 | 2.13 × 106 |
| NG50 (bp) | 2.13 × 106 | 2.13 × 106 | 2.13 × 106 | 2.13 × 106 |
| NG75 (bp) | 1.19 × 106 | 1.19 × 106 | 1.19 × 106 | 1.19 × 106 |
| Average coverage depth (x) | 115.5 | 110.0 | 42.0 | 176.0 |
| BUSCO completeness (%) | 97.4 | 98.0 | 93.9 | 98.0 |
| Short-Read Assemblies | ||||
| Mean | Median | Minimum | Maximum | |
| Total length (bp) | 3.31 × 106 | 3.30 × 106 | 3.29 × 106 | 3.47 × 106 |
| Largest contig (bp) | 4.32 × 105 | 4.17 × 105 | 3.59 × 105 | 6.10 × 105 |
| NG50 (bp) | 1.95 × 105 | 1.95 × 105 | 1.38 × 105 | 2.76 × 105 |
| NG75 (bp) | 1.18 × 105 | 1.16 × 105 | 8.06 × 104 | 1.72 × 105 |
| Average coverage depth (x) | 119.6 | 116.5 | 45.0 | 185.0 |
| BUSCO completeness (%) | 97.9 | 98.0 | 97.3 | 98.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Craddock, H.A.; Motro, Y.; Zilberman, B.; Khalfin, B.; Bardenstein, S.; Moran-Gilad, J. Long-Read Sequencing and Hybrid Assembly for Genomic Analysis of Clinical Brucella melitensis Isolates. Microorganisms 2022, 10, 619. https://doi.org/10.3390/microorganisms10030619
Craddock HA, Motro Y, Zilberman B, Khalfin B, Bardenstein S, Moran-Gilad J. Long-Read Sequencing and Hybrid Assembly for Genomic Analysis of Clinical Brucella melitensis Isolates. Microorganisms. 2022; 10(3):619. https://doi.org/10.3390/microorganisms10030619
Chicago/Turabian StyleCraddock, Hillary A., Yair Motro, Bar Zilberman, Boris Khalfin, Svetlana Bardenstein, and Jacob Moran-Gilad. 2022. "Long-Read Sequencing and Hybrid Assembly for Genomic Analysis of Clinical Brucella melitensis Isolates" Microorganisms 10, no. 3: 619. https://doi.org/10.3390/microorganisms10030619
APA StyleCraddock, H. A., Motro, Y., Zilberman, B., Khalfin, B., Bardenstein, S., & Moran-Gilad, J. (2022). Long-Read Sequencing and Hybrid Assembly for Genomic Analysis of Clinical Brucella melitensis Isolates. Microorganisms, 10(3), 619. https://doi.org/10.3390/microorganisms10030619