
Assembly and Comparison of Ca. Neoehrlichia mikurensis Genomes

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Storage
2.2. DNA Extraction, Pathogen Detection, and Enrichment
2.3. Genome Sequencing (Oxford Nanopore Technologies and Illumina)
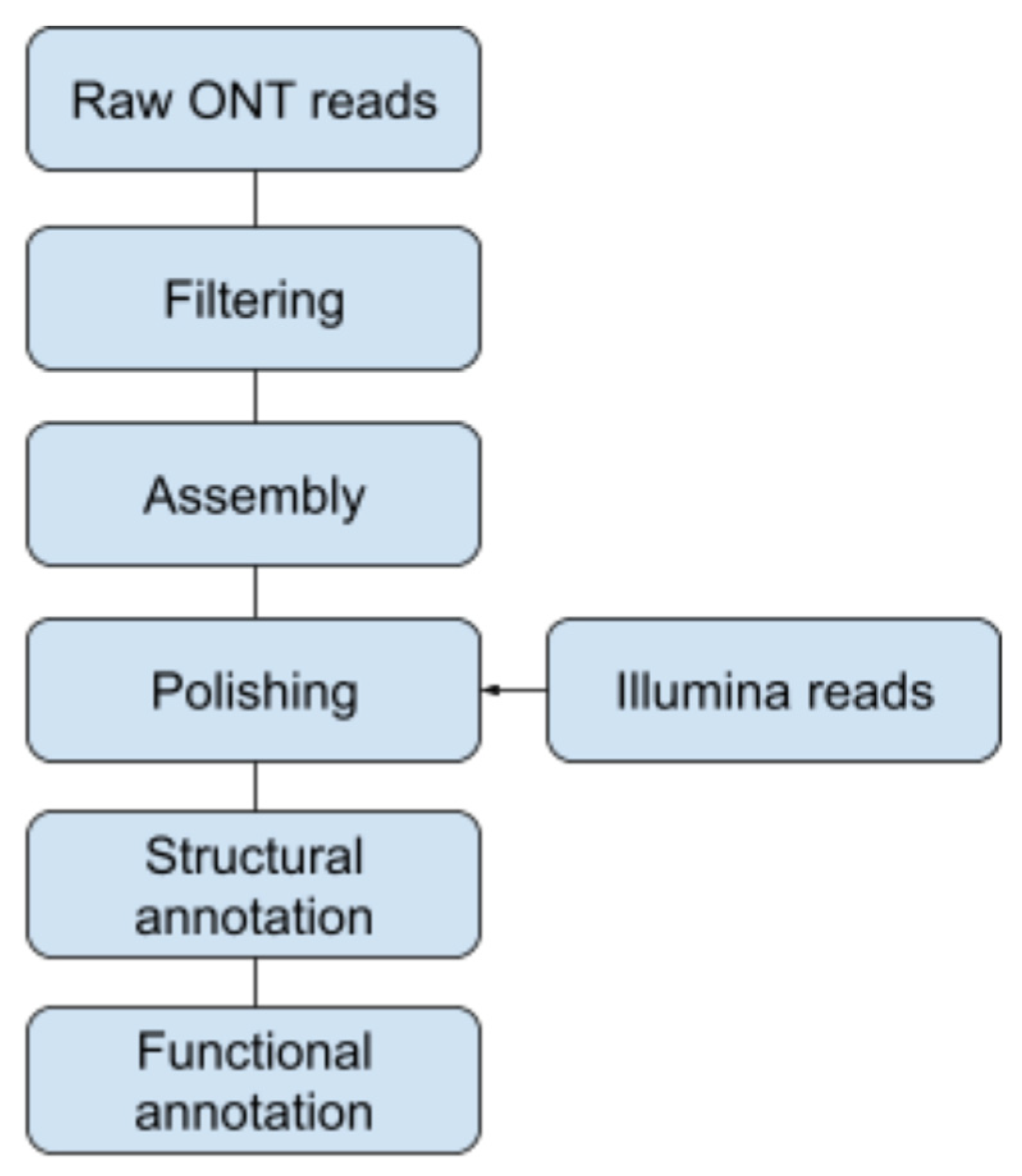
2.4. Genome Assembly and Annotation
2.5. Pangenomic and Comparative Analyses
2.6. Variant Calling
3. Results
3.1. Genomes Generated in This Study
3.2. Intraspecies Comparisons
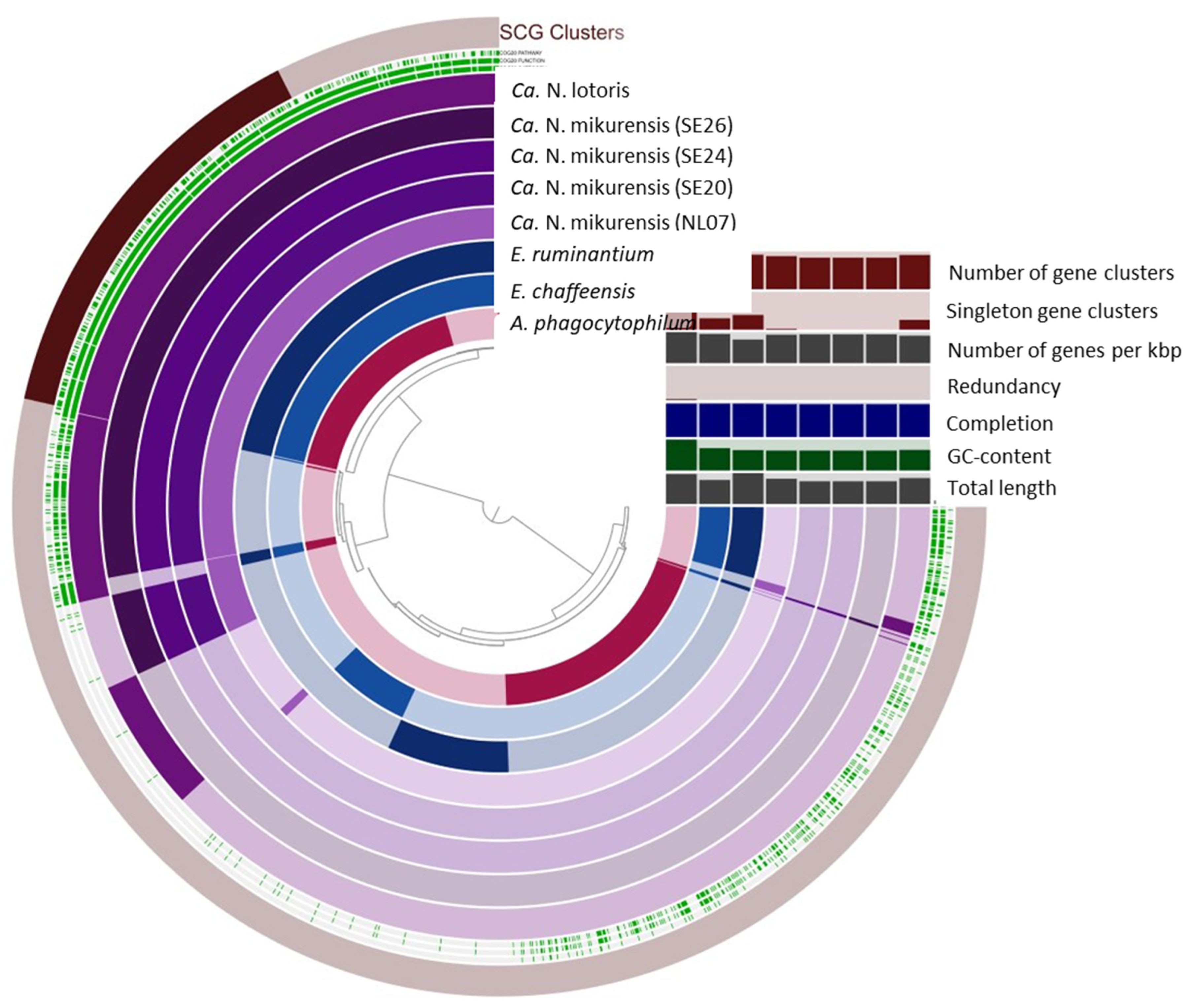
3.3. Pangenome Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Estrada-Peña, A.; Mihalca, A.D.; Petney, T.N. Ticks of Europe and North Africa: A Guide to Species Identification; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Medlock, J.M.; Hansford, K.M.; Bormane, A.; Derdakova, M.; Estrada-Pena, A.; George, J.C.; Golovljova, I.; Jaenson, T.G.; Jensen, J.K.; Jensen, P.M.; et al. Driving forces for changes in geographical distribution of Ixodes ricinus ticks in Europe. Parasites Vectors 2013, 6, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprong, H.; Azagi, T.; Hoornstra, D.; Nijhof, A.M.; Knorr, S.; Baarsma, M.E.; Hovius, J.W. Control of Lyme borreliosis and other Ixodes ricinus-borne diseases. Parasites Vectors 2018, 11, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandekerckhove, O.; De Buck, E.; Van Wijngaerden, E. Lyme disease in Western Europe: An emerging problem? A systematic review. Acta Clin. Belg. 2021, 76, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Kunze, U.; Isw, T.B.E. Tick-borne encephalitis-still on the map: Report of the 18th annual meeting of the international scientific working group on tick-borne encephalitis (ISW-TBE). Ticks Tick-Borne Dis. 2016, 7, 911–914. [Google Scholar] [CrossRef]
- Hansford, K.M.; Fonville, M.; Jahfari, S.; Sprong, H.; Medlock, J.M. Borrelia miyamotoi in host-seeking Ixodes ricinus ticks in England. Epidemiol. Infect. 2015, 143, 1079–1087. [Google Scholar] [CrossRef]
- Hansford, K.M.; Fonville, M.; Gillingham, E.L.; Coipan, E.C.; Pietzsch, M.E.; Krawczyk, A.I.; Vaux, A.G.C.; Cull, B.; Sprong, H.; Medlock, J.M. Ticks and Borrelia in urban and peri-urban green space habitats in a city in southern England. Ticks Tick-Borne Dis. 2017, 8, 353–361. [Google Scholar] [CrossRef]
- Olsthoorn, F.; Sprong, H.; Fonville, M.; Rocchi, M.; Medlock, J.; Gilbert, L.; Ghazoul, J. Occurrence of tick-borne pathogens in questing Ixodes ricinus ticks from Wester Ross, Northwest Scotland. Parasites Vectors 2021, 14, 430. [Google Scholar] [CrossRef]
- Hoper, L.; Skoog, E.; Stenson, M.; Grankvist, A.; Wass, L.; Olsen, B.; Nilsson, K.; Martensson, A.; Soderlind, J.; Sakinis, A.; et al. Vasculitis due to Candidatus Neoehrlichia mikurensis: A Cohort Study of 40 Swedish Patients. Clin. Infect. Dis. 2021, 73, e2372–e2378. [Google Scholar] [CrossRef]
- Azagi, T.; Hoornstra, D.; Kremer, K.; Hovius, J.W.R.; Sprong, H. Evaluation of Disease Causality of Rare Ixodes ricinus-Borne Infections in Europe. Pathogens 2020, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Hoornstra, D.; Harms, M.G.; Gauw, S.A.; Wagemakers, A.; Azagi, T.; Kremer, K.; Sprong, H.; van den Wijngaard, C.C.; Hovius, J.W. Ticking on Pandora’s box: A prospective case-control study into ‘other’ tick-borne diseases. BMC Infect. Dis. 2021, 21, 501. [Google Scholar] [CrossRef]
- Geebelen, L.; Lernout, T.; Tersago, K.; Terryn, S.; Hovius, J.W.; Docters van Leeuwen, A.; Van Gucht, S.; Speybroeck, N.; Sprong, H. No molecular detection of tick-borne pathogens in the blood of patients with erythema migrans in Belgium. Parasites Vectors 2022, 15, 27. [Google Scholar] [CrossRef]
- Jahfari, S.; Hofhuis, A.; Fonville, M.; van der Giessen, J.; van Pelt, W.; Sprong, H. Molecular Detection of Tick-Borne Pathogens in Humans with Tick Bites and Erythema Migrans, in the Netherlands. PLoS Negl. Trop. Dis. 2016, 10, e0005042. [Google Scholar] [CrossRef] [Green Version]
- Markowicz, M.; Schotta, A.M.; Hoss, D.; Kundi, M.; Schray, C.; Stockinger, H.; Stanek, G. Infections with Tickborne Pathogens after Tick Bite, Austria, 2015–2018. Emerg. Infect. Dis. 2021, 27, 1048. [Google Scholar] [CrossRef]
- Azagi, T.; Harms, M.; Swart, A.; Fonville, M.; Hoornstra, D.; Mughini-Gras, L.; Hovius, J.W.; Sprong, H.; van den Wijngaard, C. Self-reported symptoms and health complaints associated with exposure to Ixodes ricinus-borne pathogens. Parasites Vectors 2022, 15, 93. [Google Scholar] [CrossRef]
- Wass, L.; Grankvist, A.; Bell-Sakyi, L.; Bergstrom, M.; Ulfhammer, E.; Lingblom, C.; Wenneras, C. Cultivation of the causative agent of human neoehrlichiosis from clinical isolates identifies vascular endothelium as a target of infection. Emerg. Microbes Infect. 2019, 8, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Raoult, D. Uncultured candidatus neoehrlichia mikurensis. Clin. Infect. Dis. 2014, 59, 1042. [Google Scholar] [CrossRef]
- Grankvist, A.; Jaen-Luchoro, D.; Wass, L.; Sikora, P.; Wenneras, C. Comparative Genomics of Clinical Isolates of the Emerging Tick-Borne Pathogen Neoehrlichia mikurensis. Microorganisms 2021, 9, 1488. [Google Scholar] [CrossRef]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Quainoo, S.; Coolen, J.P.M.; van Hijum, S.; Huynen, M.A.; Melchers, W.J.G.; van Schaik, W.; Wertheim, H.F.L. Whole-Genome Sequencing of Bacterial Pathogens: The Future of Nosocomial Outbreak Analysis. Clin. Microbiol. Rev. 2017, 30, 1015–1063. [Google Scholar] [CrossRef] [Green Version]
- Pizza, M.; Scarlato, V.; Masignani, V.; Giuliani, M.M.; Arico, B.; Comanducci, M.; Jennings, G.T.; Baldi, L.; Bartolini, E.; Capecchi, B.; et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 2000, 287, 1816–1820. [Google Scholar] [CrossRef]
- Johnson, L.K.; Sahasrabudhe, R.; Gill, J.A.; Roach, J.L.; Froenicke, L.; Brown, C.T.; Whitehead, A. Draft genome assemblies using sequencing reads from Oxford Nanopore Technology and Illumina platforms for four species of North American Fundulus killifish. Gigascience 2020, 9, giaa067. [Google Scholar] [CrossRef] [PubMed]
- De Maio, N.; Shaw, L.P.; Hubbard, A.; George, S.; Sanderson, N.D.; Swann, J.; Wick, R.; AbuOun, M.; Stubberfield, E.; Hoosdally, S.J.; et al. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microb. Genom. 2019, 5, e000294. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahara, K.; Suzuki, M.; Hirabayashi, A.; Suda, W.; Hattori, M.; Suzuki, Y.; Okazaki, Y. Long-read metagenomics using PromethION uncovers oral bacteriophages and their interaction with host bacteria. Nat. Commun. 2021, 12, 27. [Google Scholar] [CrossRef]
- Neave, M.J.; Mileto, P.; Joseph, A.; Reid, T.J.; Scott, A.; Williams, D.T.; Keyburn, A.L. Comparative genomic analysis of the first Ehrlichia canis detections in Australia. Ticks Tick-Borne Dis. 2022, 13, 101909. [Google Scholar] [CrossRef]
- Liu, Z.; Peasley, A.M.; Yang, J.; Li, Y.; Guan, G.; Luo, J.; Yin, H.; Brayton, K.A. The Anaplasma ovis genome reveals a high proportion of pseudogenes. BMC Genom. 2019, 20, 69. [Google Scholar] [CrossRef] [Green Version]
- Tyler, A.D.; Mataseje, L.; Urfano, C.J.; Schmidt, L.; Antonation, K.S.; Mulvey, M.R.; Corbett, C.R. Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Sci. Rep. 2018, 8, 10931. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, M.; Ali, H.S.; Stieger, N.; Groschup, M.H.; Wolf, R.; Ulrich, R.G. Molecular identification of small mammal species using novel cytochrome B gene-derived degenerated primers. Biochem. Genet. 2012, 50, 440–447. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Bickhart, D.M.; Behsaz, B.; Gurevich, A.; Rayko, M.; Shin, S.B.; Kuhn, K.; Yuan, J.; Polevikov, E.; Smith, T.P.L.; et al. metaFlye: Scalable long-read metagenome assembly using repeat graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Medaka: Sequence Correction Provided by ONT Research. Available online: https://github.com/nanoporetech/medaka (accessed on 21 May 2021).
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Koehorst, J.J.; van Dam, J.C.J.; Saccenti, E.; Martins Dos Santos, V.A.P.; Suarez-Diez, M.; Schaap, P.J. SAPP: Functional genome annotation and analysis through a semantic framework using FAIR principles. Bioinformatics 2018, 34, 1401–1403. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Minkin, I.; Medvedev, P. Scalable multiple whole-genome alignment and locally collinear block construction with SibeliaZ. Nat. Commun. 2020, 11, 6327. [Google Scholar] [CrossRef]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet—Next generation sequence assembly visualization. Bioinformatics 2010, 26, 401–402. [Google Scholar] [CrossRef] [Green Version]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef]
- Eren, A.M.; Kiefl, E.; Shaiber, A.; Veseli, I.; Miller, S.E.; Schechter, M.S.; Fink, I.; Pan, J.N.; Yousef, M.; Fogarty, E.C.; et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 2021, 6, 3–6. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Snippy: Fast Bacterial Variant Calling from NGS Reads; GitHub, Inc.: San Francisco, CA, USA, 2015. [Google Scholar]
- Georgiades, K.; Merhej, V.; El Karkouri, K.; Raoult, D.; Pontarotti, P. Gene gain and loss events in Rickettsia and Orientia species. Biol. Direct 2011, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Andersson, S.G.; Kurland, C.G. Reductive evolution of resident genomes. Trends Microbiol. 1998, 6, 263–268. [Google Scholar] [CrossRef]
- Fournier, P.E.; El Karkouri, K.; Leroy, Q.; Robert, C.; Giumelli, B.; Renesto, P.; Socolovschi, C.; Parola, P.; Audic, S.; Raoult, D. Analysis of the Rickettsia africae genome reveals that virulence acquisition in Rickettsia species may be explained by genome reduction. BMC Genom. 2009, 10, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Karkouri, K.; Ghigo, E.; Raoult, D.; Fournier, P.E. Genomic evolution and adaptation of arthropod-associated Rickettsia. Sci. Rep. 2022, 12, 3807. [Google Scholar] [CrossRef] [PubMed]
- Rikihisa, Y. Anaplasma phagocytophilum and Ehrlichia chaffeensis: Subversive manipulators of host cells. Nat. Rev. Microbiol. 2010, 8, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Van der Woude, M.W.; Baumler, A.J. Phase and antigenic variation in bacteria. Clin. Microbiol. Rev. 2004, 17, 581–611, table of contents. [Google Scholar] [CrossRef] [Green Version]
- Dunning Hotopp, J.C.; Lin, M.; Madupu, R.; Crabtree, J.; Angiuoli, S.V.; Eisen, J.A.; Seshadri, R.; Ren, Q.; Wu, M.; Utterback, T.R.; et al. Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2006, 2, e21. [Google Scholar] [CrossRef]
- Noh, S.M.; Brayton, K.A.; Knowles, D.P.; Agnes, J.T.; Dark, M.J.; Brown, W.C.; Baszler, T.V.; Palmer, G.H. Differential expression and sequence conservation of the Anaplasma marginale msp2 gene superfamily outer membrane proteins. Infect. Immun. 2006, 74, 3471–3479. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | PromethION 18-2804 | PromethION + Illumina 18-2804 (NL07) | PromethION 18-2804 + Illumina 18-2837 (NL06) |
|---|---|---|---|
| Assembly size | 1,236,636 | 1,236,870 | 1,236,136 |
| No. CDS | 1152 | 949 | 958 |
| No. gene | 1193 | 990 | 999 |
| No. rRNA | 3 | 3 | 3 |
| No. tRNA | 37 | 37 | 37 |
| BUSCO score | 77.1% | 99.2% | 97.8% |
| Strain | Variant Total | Complex | Deletions | Insertions | SNPs | Genome Size (NL07 = 1,236,870) | % Difference |
|---|---|---|---|---|---|---|---|
| SE20 | 336 | 16 | 30 | 53 | 237 | 1,112,315 | 0.027 |
| SE24 | 349 | 13 | 31 | 48 | 257 | 1,112,301 | 0.028 |
| SE26 | 247 | 21 | 27 | 34 | 165 | 1,112,271 | 0.020 |
| Strain | Type | Nucleotide Position | Effect |
|---|---|---|---|
| SE20 | complex | 917/999 | stop_gained c.917_919delTACinsAAT p.LeuLeu306 |
| snp | 258/903 | synonymous_variant c.258C > T p.Pro86Pro | |
| snp | 792/903 | synonymous_variant c.792T > C p.Pro264Pro | |
| snp | 168/852 | synonymous_variant c.168G > A p.Pro56Pro | |
| snp | 287/852 | missense_variant c.287G > A p.Ser96Asn | |
| snp | 440/816 | missense_variant c.440C > T p.Ala147Val | |
| SE24 | complex | 917/999 | stop_gained c.917_919delTACinsAAT p.LeuLeu306 |
| snp | 792/903 | synonymous_variant c.792T > C p.Pro264Pro | |
| snp | 168/852 | synonymous_variant c.168G > A p.Pro56Pro | |
| snp | 287/852 | missense_variant c.287G > A p.Ser96Asn | |
| snp | 253/936 | missense_variant c.253C > T p.Pro85Ser | |
| SE26 | snp | 552/903 | synonymous_variant c.552A > G p.Gly184Gly |
| snp | 792/903 | synonymous_variant c.792T > C p.Pro264Pro | |
| snp | 168/852 | synonymous_variant c.168G > A p.Pro56Pro | |
| snp | 287/852 | missense_variant c.287G > A p.Ser96Asn | |
| snp | 433/816 | missense_variant c.433G > A p.Glu145Lys |
| COG Categories | Description | Number of Genes |
|---|---|---|
| L | Replication, recombination, and repair | 7 |
| J | Translation, ribosomal structure, and biogenesis | 5 |
| H | Coenzyme transport and metabolism | 3 |
| C | Energy production and conversion | 2 |
| F | Nucleotide metabolism and transport | 2 |
| M | Cell wall/membrane/envelope biogenesis | 2 |
| P | Inorganic ion transport and metabolism | 2 |
| G | Carbohydrate metabolism and transport | 1 |
| T | Signal transduction mechanisms | 1 |
| U | Intracellular trafficking, secretion, and vesicular transport | 1 |
| Microorganism | Genome Length | GC Content | Gene Clusters | Singleton Gene Clusters |
|---|---|---|---|---|
| A. phagocytophilum | 1,471,282 | 41.64 | 1018 | 523 |
| E. chaffeensis | 1,176,248 | 30.10 | 886 | 157 |
| E. ruminantium | 1,512,977 | 27.48 | 931 | 203 |
| NL07 | 1,236,870 | 26.85 | 893 | 13 |
| SE20 | 1,112,315 | 26.84 | 850 | 0 |
| SE24 | 1,112,301 | 26.84 | 850 | 0 |
| SE26 | 1,112,271 | 26.84 | 850 | 0 |
| Ca. N. lotoris | 1,268,660 | 27.75 | 923 | 135 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azagi, T.; Dirks, R.P.; Yebra-Pimentel, E.S.; Schaap, P.J.; Koehorst, J.J.; Esser, H.J.; Sprong, H. Assembly and Comparison of Ca. Neoehrlichia mikurensis Genomes. Microorganisms 2022, 10, 1134. https://doi.org/10.3390/microorganisms10061134
Azagi T, Dirks RP, Yebra-Pimentel ES, Schaap PJ, Koehorst JJ, Esser HJ, Sprong H. Assembly and Comparison of Ca. Neoehrlichia mikurensis Genomes. Microorganisms. 2022; 10(6):1134. https://doi.org/10.3390/microorganisms10061134
Chicago/Turabian StyleAzagi, Tal, Ron P. Dirks, Elena S. Yebra-Pimentel, Peter J. Schaap, Jasper J. Koehorst, Helen J. Esser, and Hein Sprong. 2022. "Assembly and Comparison of Ca. Neoehrlichia mikurensis Genomes" Microorganisms 10, no. 6: 1134. https://doi.org/10.3390/microorganisms10061134
APA StyleAzagi, T., Dirks, R. P., Yebra-Pimentel, E. S., Schaap, P. J., Koehorst, J. J., Esser, H. J., & Sprong, H. (2022). Assembly and Comparison of Ca. Neoehrlichia mikurensis Genomes. Microorganisms, 10(6), 1134. https://doi.org/10.3390/microorganisms10061134