
The Association between Gut Microbiome Diversity and Composition and Heat Tolerance in Cattle

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Fecal Sampling
2.3. 16S rRNA Sequencing
2.4. Sequence Data Processing
2.5. Correlation Analysis and Differential Feature Selection
2.6. Statistical Analysis
3. Results
3.1. Quality Assessment of Sequencing Data and ASV Analysis
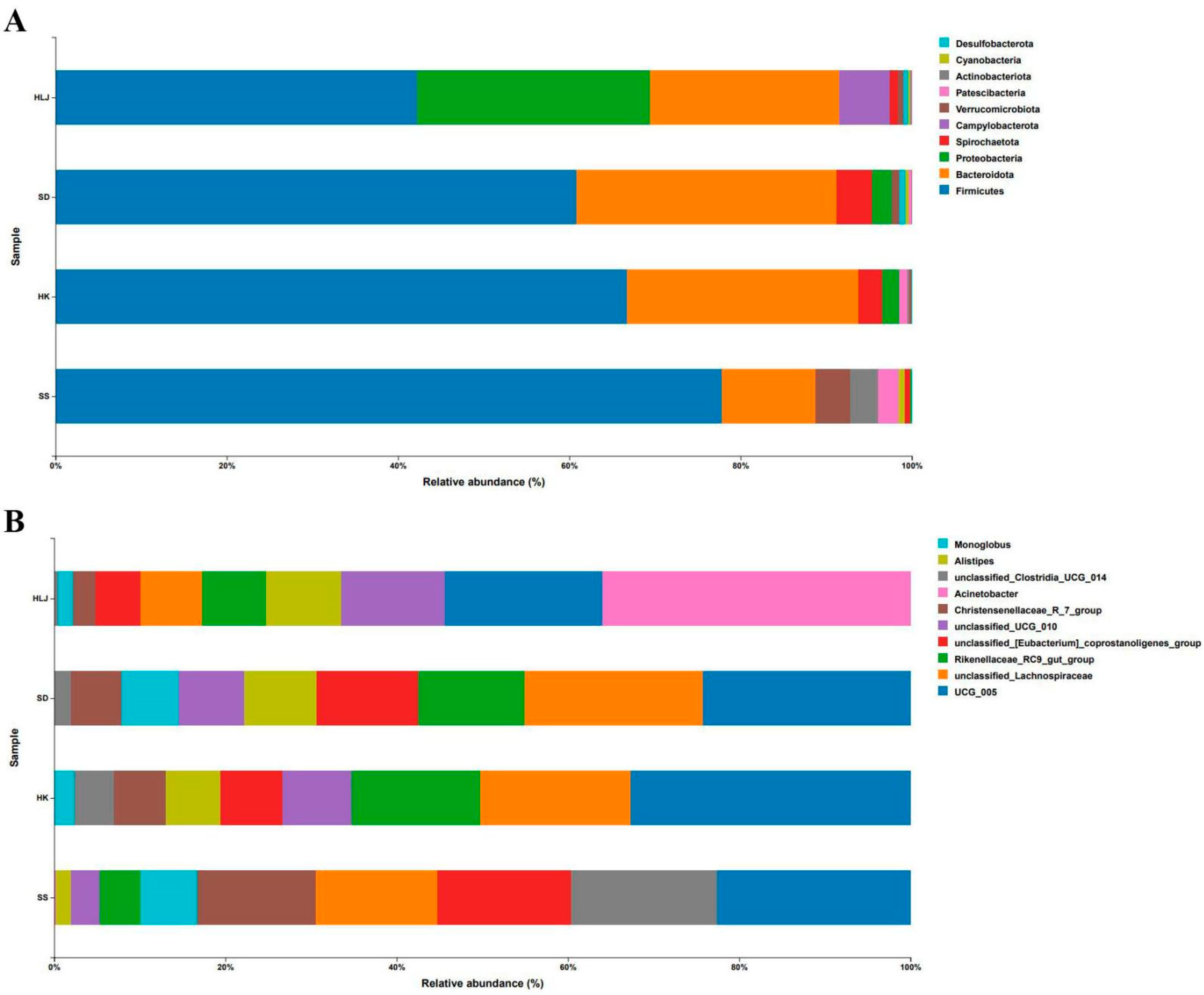
3.2. Taxonomy Analysis
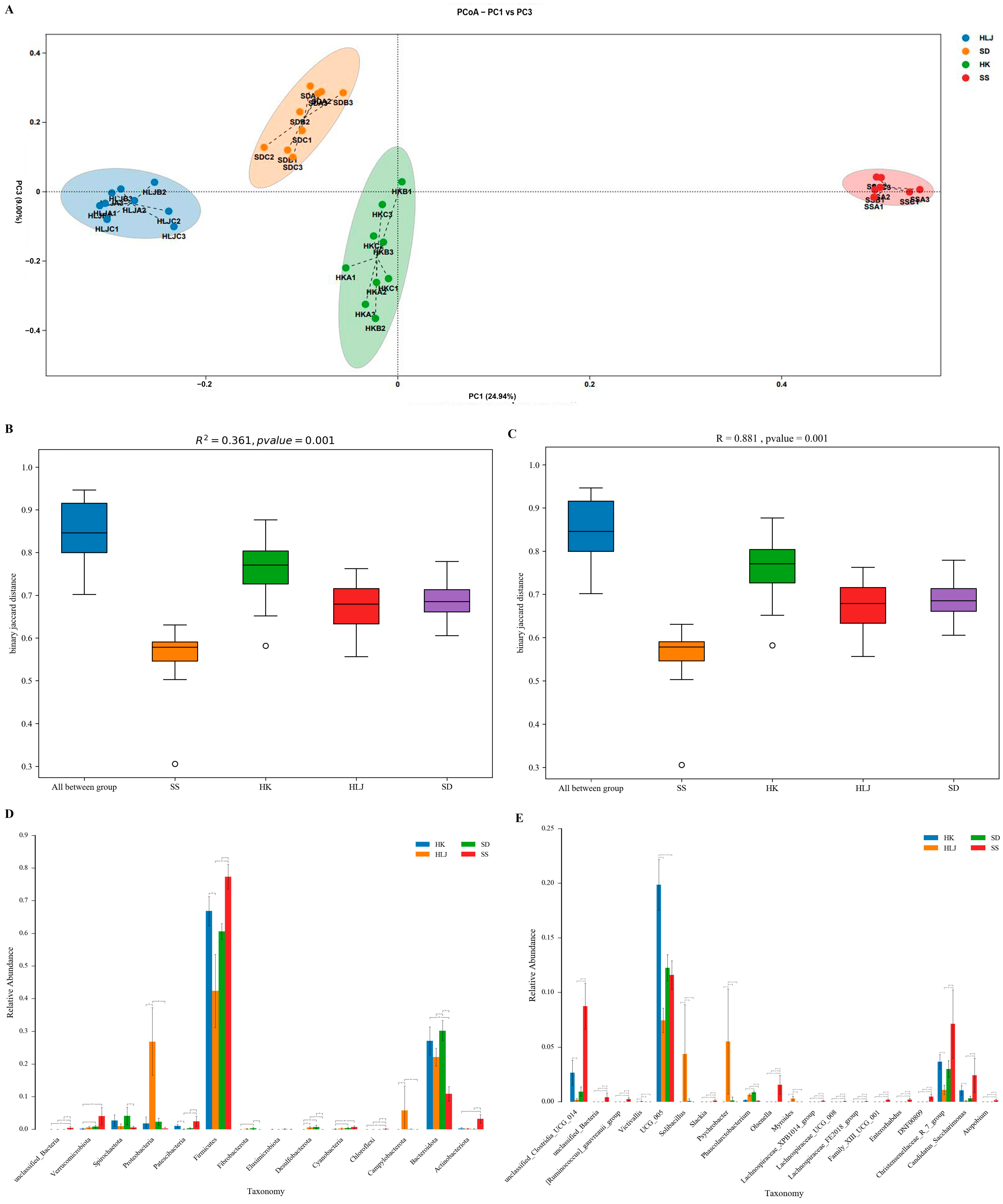
3.3. No Significant Difference Was Observed in Microbiota Diversity among the Cattle from the Same Location
3.4. The Microbiota Diversity of Cattle Displayed an Obvious Association with Geographical and Climatic Features
3.5. There Were Biomarkers to Match the Geographical and Climatic Characteristics
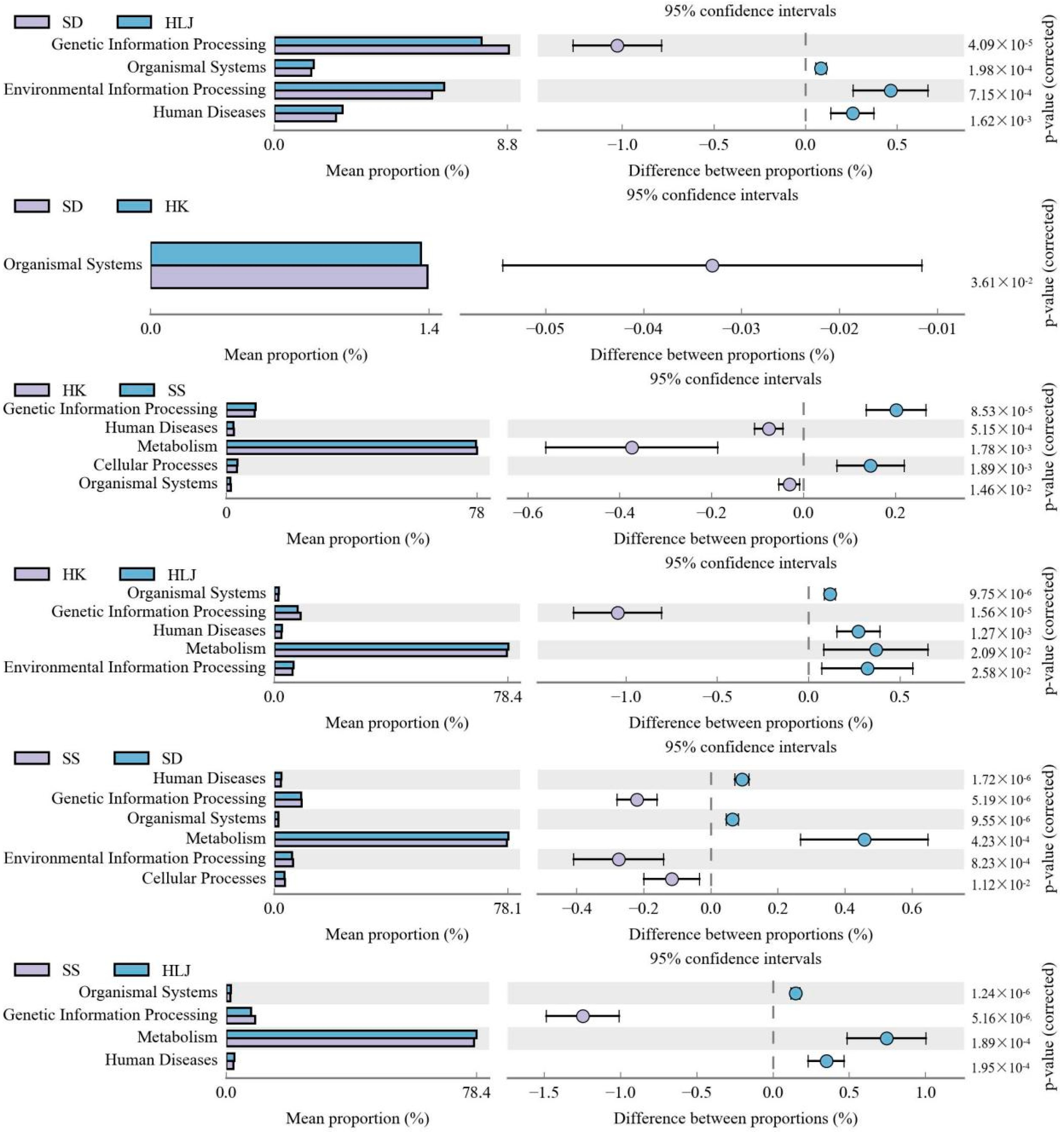
3.6. Functional Differences in Microbiota in Different Geographical and Climatic Areas
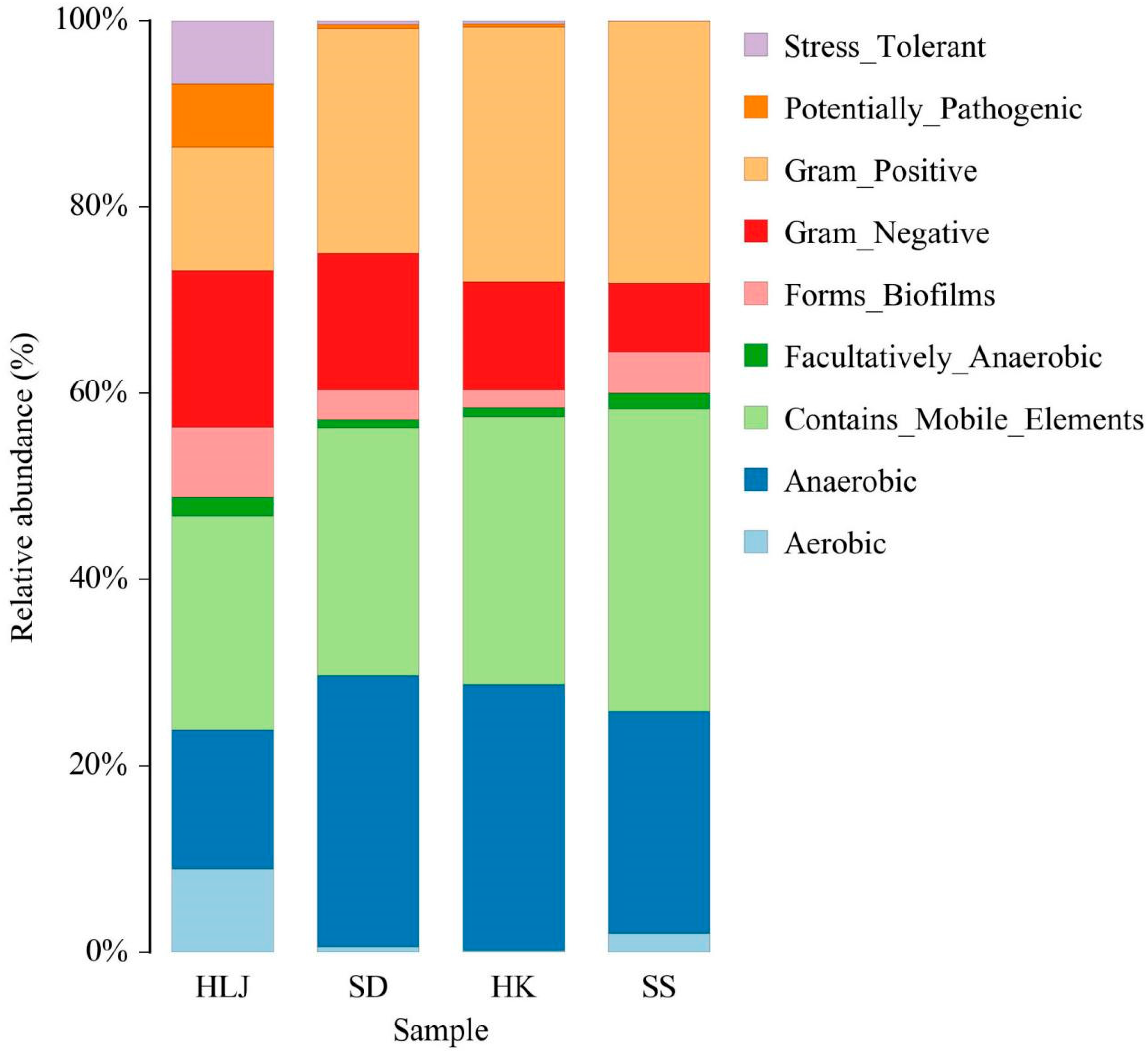
3.7. Some Microbial Phenotypes at the Organism Level Were Matched to the Geographical and Climatic Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carvajal, M.A.; Alaniz, A.J.; Gutiérrez-Gómez, C.; Vergara, P.M.; Sejian, V.; Bozinovic, F. Increasing importance of heat stress for cattle farming under future global climate scenarios. Sci. Total Environ. 2021, 801, 149661. [Google Scholar] [CrossRef]
- De Rensis, F.; Marconi, P.; Capelli, T.; Gatti, F.; Facciolongo, F.; Franzini, S.; Scaramuzzi, R.J. Fertility in postpartum dairy cows in winter or summer following estrus synchronization and fixed time AI after the induction of an LH surge with GnRH or hCG. Theriogenology 2002, 58, 1675–1687. [Google Scholar]
- Mishra, S.R. Significance of molecular chaperones and micro RNAs in acquisition of thermo-tolerance in dairy cattle. Anim. Biotechnol. 2020, 29, 1–11. [Google Scholar]
- Wang, Y.; Yang, C.; Elsheikh, N.A.H.; Li, C.; Yang, F.; Wang, G.; Li, L. HO-1 reduces heat stress-induced apoptosis in bovine granulosa cells by suppressing oxidative stress. Aging 2019, 11, 5535–5547. [Google Scholar] [CrossRef]
- Correia Sales, G.F.; Carvalho, B.F.; Schwan, R.F.; de Figueiredo Vilela, L.; Moreno Meneses, J.A.; Gionbelli, M.P.; da Silva Ávila, C.L. Heat stress influence the microbiota and organic acids concentration in beef cattle rumen. J. Therm. Biol. 2021, 97, 102897. [Google Scholar]
- Mishra, S.R. Behavioural, physiological, neuro-endocrine and molecular responses of cattle against heat stress: An updated review. Trop. Anim. Health Prod. 2021, 53, 400. [Google Scholar]
- Lu, X.; Arbab, A.A.I.; Zhang, Z.; Fan, Y.; Han, Z.; Gao, Q.; Sun, Y.; Yang, Z. Comparative Transcriptomic Analysis of the Pituitary Gland between Cattle Breeds Differing in Growth: Yunling Cattle and Leiqiong Cattle. Animals 2020, 10, 1271. [Google Scholar] [CrossRef]
- Kong, L.; Liu, G.; Deng, M.; Lian, Z.; Han, Y.; Sun, B.; Guo, Y.; Liu, D.; Li, Y. Growth retardation-responsive analysis of mRNAs and long noncoding RNAs in the liver tissue of Leiqiong cattle. Sci. Rep. 2020, 10, 14254. [Google Scholar] [CrossRef]
- Ahmad, M.; Bhatti, J.A.; Abdullah, M.; Javed, K.; Ali, M.; Rashid, G.; Uddin, R.; Badini, A.H.; Jehan, M. Effect of ambient management interventions on the production and physiological performance of lactating Sahiwal cattle during hot dry summer. Trop. Anim. Health Prod. 2018, 50, 1249–1254. [Google Scholar] [CrossRef]
- Chen, S.; Wang, J.; Peng, D.; Li, G.; Chen, J.; Gu, X. Exposure to heat-stress environment affects the physiology, circulation levels of cytokines, and microbiome in dairy cows. Sci. Rep. 2018, 8, 14606. [Google Scholar] [CrossRef]
- Planer, J.D.; Peng, Y.; Kau, A.L.; Blanton, L.V.; Ndao, I.M.; Tarr, P.I.; Warner, B.B.; Gordon, J.I. Development of the gut microbiota and mucosal IgA responses in twins and gnotobiotic mice. Nature 2016, 534, 263–266. [Google Scholar] [CrossRef]
- Lee, W.-J.; Hase, K. Gut microbiota–generated metabolites in animal health and disease. Nat. Chem. Biol. 2014, 10, 416–424. [Google Scholar] [CrossRef]
- Zhao, L.; Li, X.; Atwill, E.R.; Aly, S.S.; Williams, D.R.; Su, Z. Dynamic changes in fecal bacterial microbiota of dairy cattle across the production line. BMC Microbiol. 2022, 22, 132. [Google Scholar] [CrossRef]
- Gabanyi, I.; Lepousez, G.; Wheeler, R.; Vieites-Prado, A.; Nissant, A.; Wagner, S.; Moigneu, C.; Dulauroy, S.; Hicham, S.; Polomack, B.; et al. Bacterial sensing via neuronal Nod2 regulates appetite and body temperature. Science 2022, 376, eabj3986. [Google Scholar] [CrossRef]
- Louca, P.; Nogal, A.; Wells, P.M.; Asnicar, F.; Wolf, J.; Steves, C.J.; Spector, T.D.; Segata, N.; Berry, S.E.; Valdes, A.M.; et al. Gut microbiome diversity and composition is associated with hypertension in women. J. Hypertens. 2021, 39, 1810–1816. [Google Scholar] [CrossRef]
- Lan, D.; Jiangjiang, Z.; Lin, B.; Chen, Y.; Huang, C.; Xiong, X.; Fu, M.; Mipam, T.D.; Ai, Y.; Zeng, B.; et al. Correlations between gut microbiota community structures of Tibetans and geography. Sci. Rep. 2017, 7, 16982. [Google Scholar] [CrossRef]
- Zhao, J.; Yao, Y.; Li, D.; Xu, H.; Wu, J.; Wen, A.; Xie, M.; Ni, Q.; Zhang, M.; Peng, G.; et al. Characterization of the Gut Microbiota in Six Geographical Populations of Chinese Rhesus Macaques (Macaca mulatta), Implying an Adaptation to High-Altitude Environment. Microb. Ecol. 2018, 76, 565–577. [Google Scholar] [CrossRef]
- Goel, A.; Kim, B.-J.; Ncho, C.-M.; Jeong, C.-M.; Gupta, V.; Jung, J.-Y.; Ha, S.-Y.; Lee, D.-H.; Yang, J.-K.; Choi, Y.-H. Dietary Supplementation of Shredded, Steam-Exploded Pine Particles Decreases Pathogenic Microbes in the Cecum of Acute Heat-Stressed Broilers. Animals 2021, 11, 2252. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, J.; Zhou, M.; Yin, P.; Chen, Z.; Zhao, Q.; Hu, K.; Liu, Q.; Ou, C.-Q. Hourly temperature variability and mortality in 31 major Chinese cities: Effect modification by individual characteristics, season and temperature zone. Environ. Int. 2021, 156, 106746. [Google Scholar] [CrossRef]
- Ding, D.; Zhu, J.; Gao, Y.; Yang, F.; Ma, Y.; Cheng, X.; Li, J.; Dong, P.; Yang, H.; Chen, S. Effect of cattle farm exposure on oropharyngeal and gut microbial communities and antibiotic resistance genes in workers. Sci. Total Environ. 2022, 806, 150685. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Ward, T.; Larson, J.; Meulemans, J.; Hillmann, B.; Lynch, J.; Sidiropoulos, D.; Spear, J.R.; Caporaso, G.; Blekhman, R.; Knight, R.; et al. BugBase predicts organism-level microbiome phenotypes. bioRxiv 2017, 133462. [Google Scholar] [CrossRef]
- MacPherson, A.J.; Harris, N.L. Interactions between commensal intestinal bacteria and the immune system. Nat. Rev. Immunol. 2004, 4, 478–485. [Google Scholar] [CrossRef]
- Xu, Q.; Qiao, Q.; Gao, Y.; Hou, J.; Hu, M.; Du, Y.; Zhao, K.; Li, X. Gut Microbiota and Their Role in Health and Metabolic Disease of Dairy Cow. Front. Nutr. 2021, 8, 701511. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.; Wang, L.; Xue, B.; Hu, R.; Zou, H.; Liu, S.; Shah, A.M.; Peng, Q. Comparison of changes in fecal microbiota of calves with and without dam. PeerJ 2022, 10, e12826. [Google Scholar] [CrossRef]
- Corrêa, P.S.; Jimenez, C.R.; Mendes, L.W.; Rymer, C.; Ray, P.; Gerdes, L.; da Silva, V.O.; De Nadai Fernandes, E.A.; Abdalla, A.L.; Louvandini, H. Taxonomy and Functional Diversity in the Fecal Microbiome of Beef Cattle Reared in Brazilian Traditional and Semi-Intensive Production Systems. Front. Microbiol. 2021, 12, 768480. [Google Scholar] [CrossRef]
- Redding, L.E.; Berry, A.S.; Indugu, N.; Huang, E.; Beiting, D.P.; Pitta, D. Gut microbiota features associated with Clostridioides difficile colonization in dairy calves. PLoS ONE 2021, 16, e0251999. [Google Scholar] [CrossRef]
- Dill-McFarland, K.A.; Breaker, J.D.; Suen, G. Microbial succession in the gastrointestinal tract of dairy cows from 2 weeks to first lactation. Sci. Rep. 2017, 7, 40864. [Google Scholar] [CrossRef]
- Dill-McFarland, K.A.; Weimer, P.J.; Breaker, J.D.; Suen, G. Diet Influences Early Microbiota Development in Dairy Calves without Long-Term Impacts on Milk Production. Appl. Environ. Microbiol. 2019, 85, e02141-18. [Google Scholar] [CrossRef]
- Klein-Jöbstl, D.; Schornsteiner, E.; Mann, E.; Wagner, M.; Drillich, M.; Schmitz-Esser, S. Pyrosequencing reveals diverse fecal mi-crobiota in Simmental calves during early development. Front. Microbiol. 2014, 5, 622. [Google Scholar]
- Meale, S.J.; Li, S.; Azevedo, P.; Derakhshani, H.; Plaizier, J.C.; Khafipour, E.; Steele, M.A. Development of Ruminal and Fecal Micro-biomes Are Affected by Weaning but Not Weaning Strategy in Dairy Calves. Front. Microbiol. 2016, 7, 582. [Google Scholar]
- Díaz Carrasco, J.M.; Cabral, C.; Redondo, L.M.; Pin Viso, N.D.; Colombatto, D.; Farber, M.D.; Fernández Miyakawa, M.E. Impact of Chestnut and Quebracho Tannins on Rumen Microbiota of Bovines. Biomed. Res. Int. 2017, 2017, 9610810. [Google Scholar]
- Jami, E.; White, B.A.; Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed ef-ficiency. PLoS ONE 2014, 9, e85423. [Google Scholar]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The met-agenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 2008, 9, 386. [Google Scholar]
- Zhang, X.; Chen, B.; Wu, J.; Sha, J.; Yang, B.; Zhu, J.; Sun, J.; Hartung, J.; Bao, E. Aspirin Enhances the Protection of Hsp90 from Heat-Stressed Injury in Cardiac Microvascular Endothelial Cells Through PI3K-Akt and PKM2 Pathways. Cells 2020, 9, 243. [Google Scholar] [CrossRef]
- Chen, H.; Adam, A.; Cheng, Y.; Tang, S.; Hartung, J.; Bao, E. Localization and expression of heat shock protein 70 with rat myocardial cell damage induced by heat stress in vitro and in vivo. Mol. Med. Rep. 2015, 11, 2276–2284. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Li, L.; Zhao, X. Comparative analyses of fecal microbiota in Tibetan and Chinese Han living at low or high altitude by barcoded 454 pyrosequencing. Sci. Rep. 2015, 5, 14682. [Google Scholar] [CrossRef]
- Watanabe, M.; Kojima, H.; Fukui, M. Desulfotomaculum intricatum sp. nov., a sulfate reducer isolated from freshwater lake sediment. Int. J. Syst. Evol. Microbiol. 2013, 63, 3574–3578. [Google Scholar] [CrossRef]
- Geerlings, S.Y.; Kostopoulos, I.; De Vos, W.M.; Belzer, C. Akkermansia muciniphila in the Human Gastrointestinal Tract: When, Where, and How? Microorganisms 2018, 6, 75. [Google Scholar] [CrossRef]
- Rizzatti, G.; Lopetuso, L.R.; Gibiino, G.; Binda, C.; Gasbarrini, A. Proteobacteria: A Common Factor in Human Diseases. BioMed. Res. Int. 2017, 2017, 9351507. [Google Scholar] [CrossRef]
- Auffret, M.D.; Dewhurst, R.J.; Duthie, C.-A.; Rooke, J.A.; Wallace, R.J.; Freeman, T.C.; Stewart, R.; Watson, M.; Roehe, R. The rumen microbiome as a reservoir of antimicrobial resistance and pathogenicity genes is directly affected by diet in beef cattle. Microbiome 2017, 5, 159. [Google Scholar] [CrossRef]
- Wu, R.; Mei, X.; Wang, J.; Sun, W.; Xue, T.; Lin, C.; Xu, D. Zn(ii)-Curcumin supplementation alleviates gut dysbiosis and zinc dyshomeostasis during doxorubicin-induced cardiotoxicity in rats. Food Funct. 2019, 10, 5587–5604. [Google Scholar] [CrossRef]
- Hernández-Álvarez, C.; García-Oliva, F.; Cruz-Ortega, R.; Romero, M.F.; Barajas, H.R.; Piñero, D.; Alcaraz, L.D. Squash root microbiome transplants and metagenomic inspection for in situ arid adaptations. Sci. Total Environ. 2021, 805, 150136. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, C.; Li, W.; Weng, S.; Song, W.; Li, J.; Wang, Y. Functional diversity of microbial communities in inactive seafloor sulfide deposits. FEMS Microbiol. Ecol. 2021, 97, fiab108. [Google Scholar] [CrossRef]
- Kikuchi, H.; Watanabe, T.; Jia, Z.; Kimura, M.; Asakawa, S. Molecular analyses reveal stability of bacterial communities in bulk soil of a Japanese paddy field: Estimation by denaturing gradient gel electrophoresis of 16S rRNA genes amplified from DNA accompanied with RNA. Soil Sci. Plant Nutr. 2007, 53, 448–458. [Google Scholar] [CrossRef]
- Man, S.M. The clinical importance of emerging Campylobacter species. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 669–685. [Google Scholar] [CrossRef]
- Zeigler, C.C.; Persson, G.R.; Wondimu, B.; Marcus, C.; Sobko, T.; Modéer, T. Microbiota in the Oral Subgingival Biofilm Is Associated with Obesity in Adolescence. Obesity 2012, 20, 157–164. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, Y.; Yin, S.; Zhang, S.; Wang, L.; Qu, Y. Effect of acidified milk feeding on the intake, average daily gain and fecal microbiological diversity of Holstein dairy calves. Asian-Australas. J. Anim. Sci. 2020, 33, 1265–1272. [Google Scholar]
- Ho, T.T.B.; Groer, M.W.; Kane, B.; Yee, A.L.; Torres, B.A.; Gilbert, J.A.; Maheshwari, A. Dichotomous development of the gut microbiome in preterm infants. Microbiome 2018, 6, 157. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, H.; Xu, T.; Hu, Y.; Jiang, Y. Acute exposure to simulated high-altitude hypoxia alters gut microbiota in mice. Arch. Microbiol. 2022, 204, 412. [Google Scholar]
- Drenkard, E. Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 2003, 5, 1213–1219. [Google Scholar] [CrossRef]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial biofilm and associated infections. J. Chin. Med. Assoc. 2018, 81, 7–11. [Google Scholar] [CrossRef]
- Murray, A.J.; Montgomery, H.E.; Feelisch, M.; Grocott, M.P.W.; Martin, D.S. Metabolic adjustment to high-altitude hypoxia: From genetic signals to physiological implications. Biochem. Soc. Trans. 2018, 46, 599–607. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age Samples No. Location | HLJ | SD | HK | SS |
|---|---|---|---|---|
| 4–6 years old (A) | 3 | 3 | 3 | 3 |
| 1.5–2 years old (B) | 3 | 3 | 3 | 1 |
| 6 months to 1 year old (C) | 3 | 3 | 3 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Cui, K.; Wen, X.; Li, L.; Yu, X.; Li, B.; Lin, H.; He, H.; Wang, F. The Association between Gut Microbiome Diversity and Composition and Heat Tolerance in Cattle. Microorganisms 2022, 10, 1672. https://doi.org/10.3390/microorganisms10081672
Zhang X, Cui K, Wen X, Li L, Yu X, Li B, Lin H, He H, Wang F. The Association between Gut Microbiome Diversity and Composition and Heat Tolerance in Cattle. Microorganisms. 2022; 10(8):1672. https://doi.org/10.3390/microorganisms10081672
Chicago/Turabian StyleZhang, Xiaohui, Ke Cui, Xiaobo Wen, Lianbin Li, Xiangchun Yu, Boling Li, Haichao Lin, Hongxuan He, and Fengyang Wang. 2022. "The Association between Gut Microbiome Diversity and Composition and Heat Tolerance in Cattle" Microorganisms 10, no. 8: 1672. https://doi.org/10.3390/microorganisms10081672