
How to Shut Down Transcription in Archaea during Virus Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Archaeal Transcription Machinery
1.2. Viruses versus Hosts
1.3. Virus-Encoded Inhibitors
1.4. Cellular Inhibitors
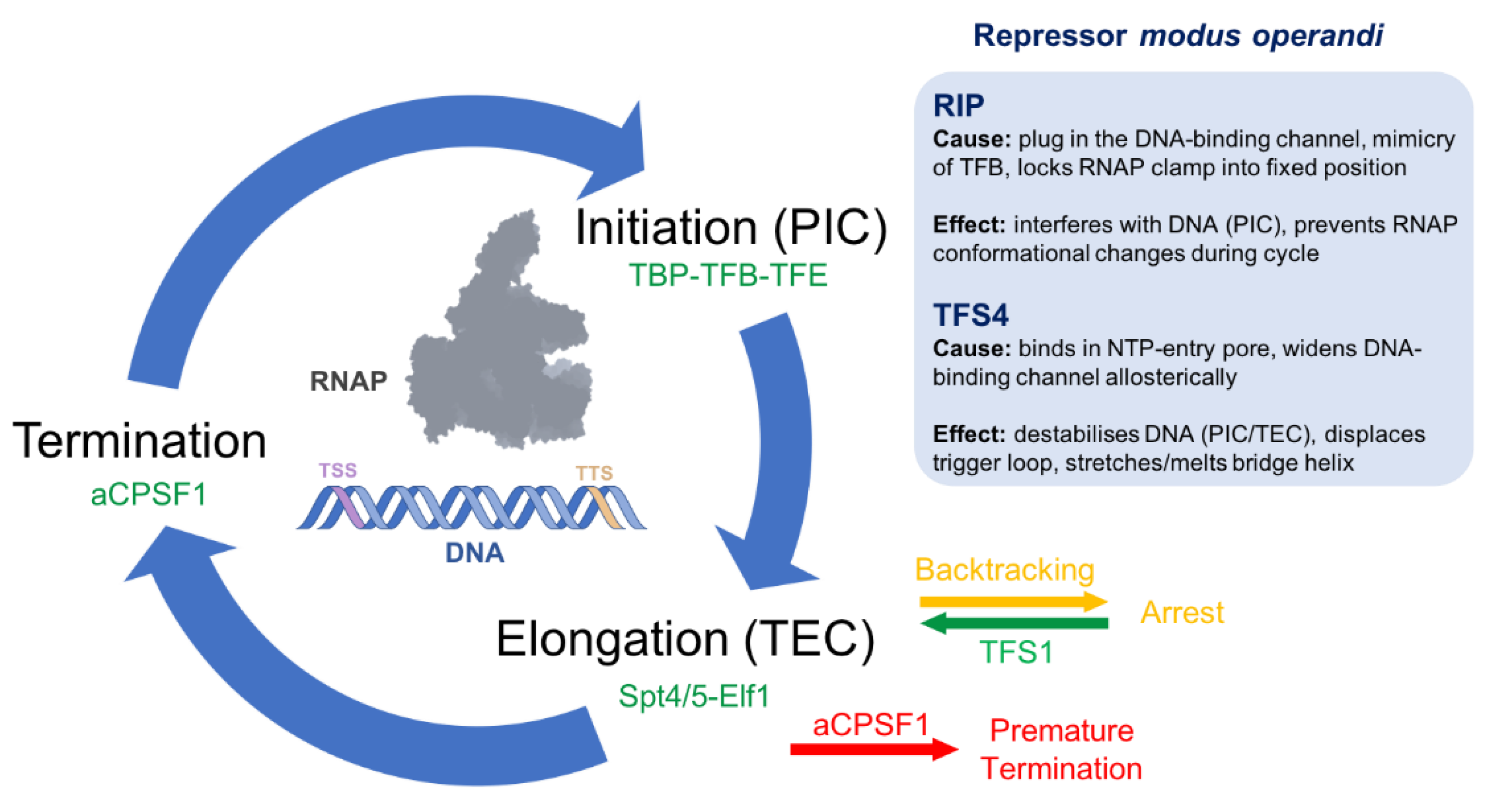
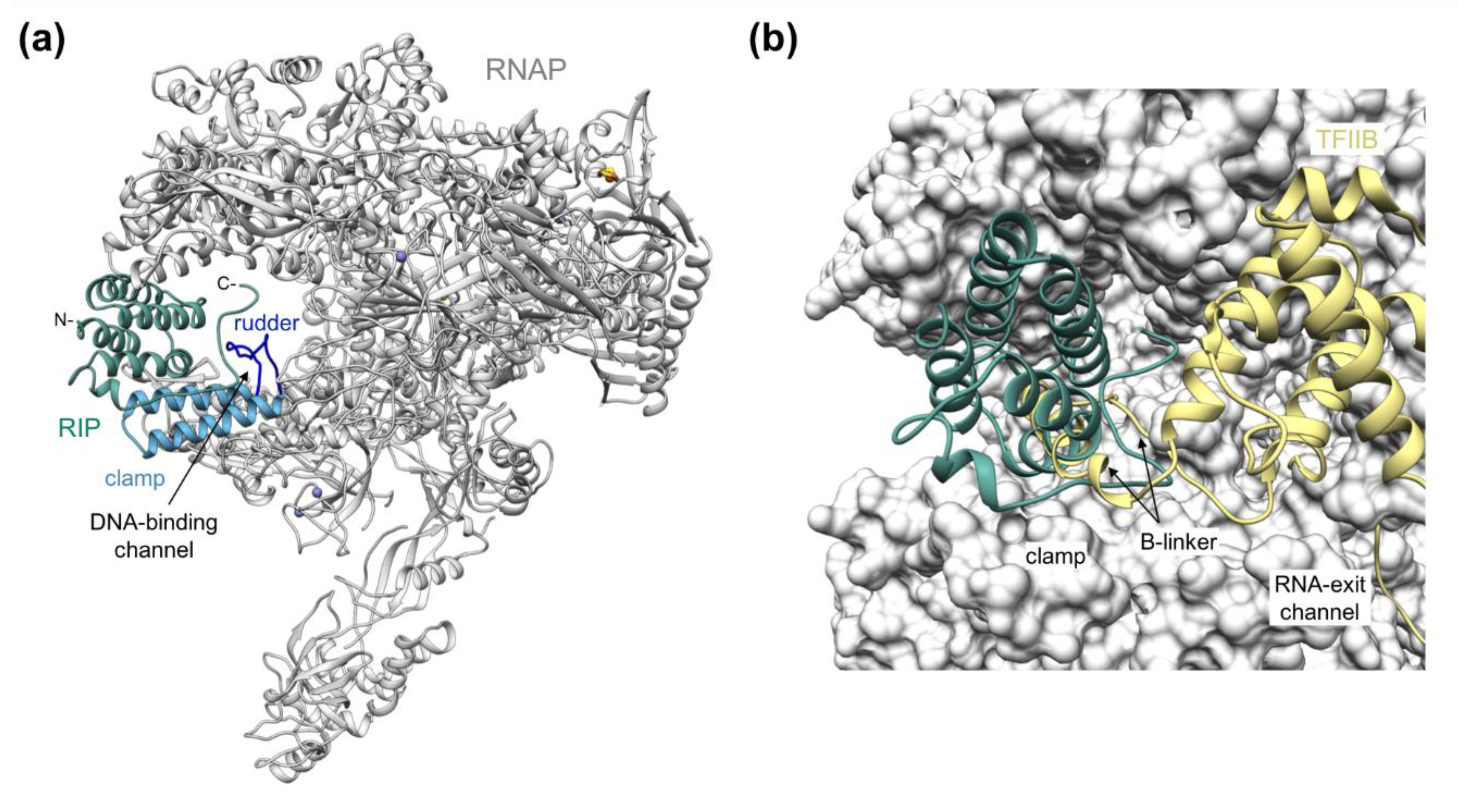
2. RIP Forms a Plug in the DNA-Binding Channel
3. TFS4 Uses a ‘Belts and Braces’ Approach to Inhibition
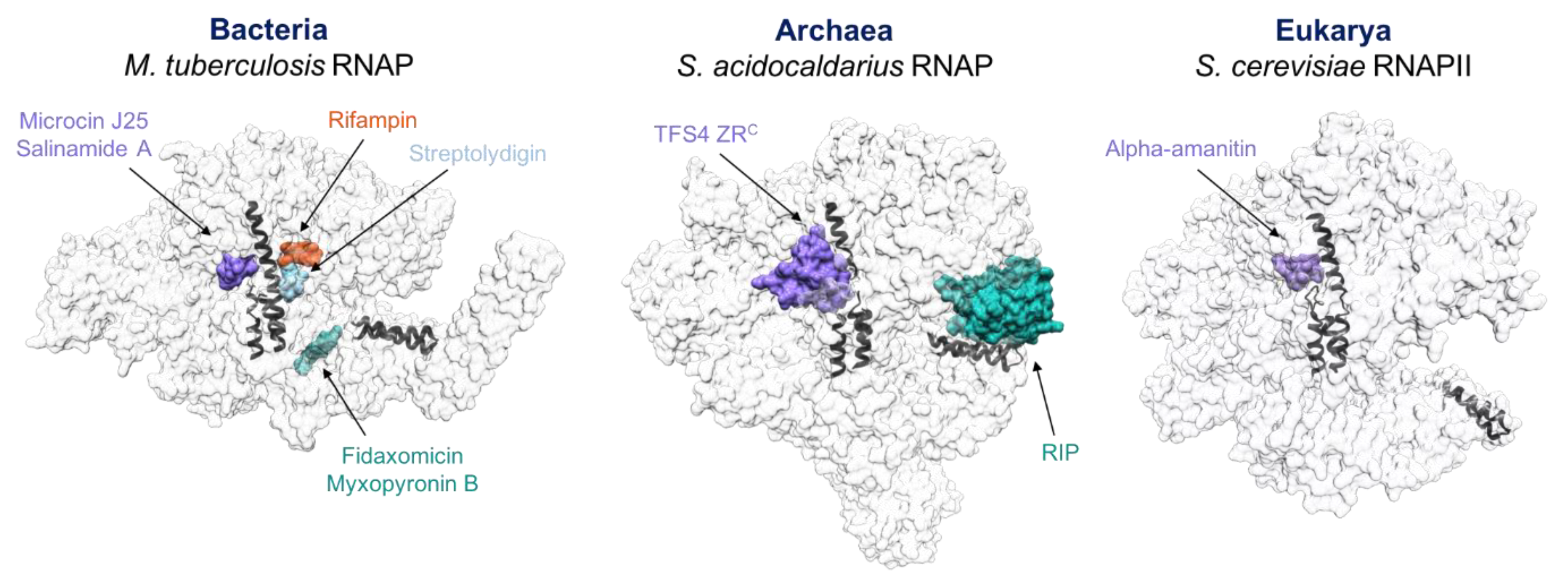
4. Similarities between Antibiotics, Inhibitors, TFS4 and RIP
5. Discussion
Take Home Messages
- RNAPs and the molecular mechanisms of RNA synthesis are universally conserved in all domains of life.
- Both the virus and host cell can encode repressors that tightly bind to RNAPs and efficiently inhibit their functions, leading to transcriptome repression or attenuation.
- The modus operandi of these factors can unravel the underlying molecular mechanisms of RNAP activity and identify critical pressure points of enzyme function
- Inhibitory mechanisms include:
- ○
- steric occlusion of DNA, NTPs and transcription factor binding sites;
- ○
- allosteric regulation by inducing conformational changes that perturb the active site (bridge helix and trigger loop) and widening of the DNA-binding channel.
- Allosteric mechanisms of inhibition can reveal movements inherent to RNAP function. As these are evolutionary conserved, it is possible to draw intriguing parallels between the regulation of different transcription systems.
- Antibiotics that target and inhibit RNAPs and proteinaceous repressors act via functionally closely related molecular mechanisms.
- A thorough understanding of RNAP inhibition in all domains of life, including archaea, could be beneficial for the development of novel antibiotics.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Offre, P.; Spang, A.; Schleper, C. Archaea in biogeochemical cycles. Annu. Rev. Microbiol. 2013, 67, 437–457. [Google Scholar] [CrossRef] [PubMed]
- Hoegenauer, C.; Hammer, H.F.; Mahnert, A.; Moissl-Eichinger, C. Methanogenic archaea in the human gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Dame-Teixeira, N.; de Cena, J.A.; Cortes, D.A.; Belmok, A.; Dos Anjos Borges, L.G.; Marconatto, L.; Giongo, A.; Kyaw, C.M. Presence of Archaea in dental caries biofilms. Arch. Oral Biol. 2020, 110, 104606. [Google Scholar] [CrossRef]
- Bang, C.; Schmitz, R.A. Archaea associated with human surfaces: Not to be underestimated. FEMS Microbiol. Rev. 2015, 39, 631–648. [Google Scholar] [CrossRef]
- Eme, L.; Spang, A.; Lombard, J.; Stairs, C.W.; Ettema, T.J.G. Archaea and the origin of eukaryotes. Nat. Rev. Microbiol. 2018, 16, 120. [Google Scholar] [CrossRef]
- Peeters, E.; Driessen, R.P.; Werner, F.; Dame, R.T. The interplay between nucleoid organization and transcription in archaeal genomes. Nat. Rev. Microbiol. 2015, 13, 333–341. [Google Scholar] [CrossRef]
- Werner, F.; Grohmann, D. Evolution of multisubunit RNA polymerases in the three domains of life. Nat. Rev. Microbiol. 2011, 9, 85–98. [Google Scholar] [CrossRef]
- Mattiroli, F.; Bhattacharyya, S.; Dyer, P.N.; White, A.E.; Sandman, K.; Burkhart, B.W.; Byrne, K.R.; Lee, T.; Ahn, N.G.; Santangelo, T.J.; et al. Structure of histone-based chromatin in Archaea. Science 2017, 357, 609–612. [Google Scholar] [CrossRef]
- Fouqueau, T.; Blombach, F.; Cackett, G.; Carty, A.E.; Matelska, D.M.; Ofer, S.; Pilotto, S.; Phung, D.K.; Werner, F. The cutting edge of archaeal transcription. Emerg. Top. Life Sci. 2018, 2, 517–533. [Google Scholar] [CrossRef]
- Fouqueau, T.; Blombach, F.; Hartman, R.; Cheung, A.C.M.; Young, M.J.; Werner, F. The transcript cleavage factor paralogue TFS4 is a potent RNA polymerase inhibitor. Nat. Commun. 2017, 8, 1914. [Google Scholar] [CrossRef] [Green Version]
- Blombach, F.; Matelska, D.; Fouqueau, T.; Cackett, G.; Werner, F. Key Concepts and Challenges in Archaeal Transcription. J. Mol. Biol. 2019, 431, 4184–4201. [Google Scholar] [CrossRef] [PubMed]
- Blombach, F.; Fouqueau, T.; Matelska, D.; Smollett, K.; Werner, F. Promoter-proximal elongation regulates transcription in archaea. Nat. Commun. 2021, 12, 5524. [Google Scholar] [CrossRef] [PubMed]
- Smollett, K.; Blombach, F.; Reichelt, R.; Thomm, M.; Werner, F. A global analysis of transcription reveals two modes of Spt4/5 recruitment to archaeal RNA polymerase. Nat. Microbiol. 2017, 2, 17021. [Google Scholar] [CrossRef]
- Parvin, J.D.; Sharp, P.A. DNA topology and a minimal set of basal factors for transcription by RNA polymerase II. Cell 1993, 73, 533–540. [Google Scholar] [CrossRef]
- Blombach, F.; Salvadori, E.; Fouqueau, T.; Yan, J.; Reimann, J.; Sheppard, C.; Smollett, K.L.; Albers, S.V.; Kay, C.W.; Thalassinos, K.; et al. Archaeal TFEalpha/beta is a hybrid of TFIIE and the RNA polymerase III subcomplex hRPC62/39. eLife 2015, 4, e08378. [Google Scholar] [CrossRef] [PubMed]
- Fouqueau, T.; Werner, F. The architecture of transcription elongation. Science 2017, 357, 871–872. [Google Scholar] [CrossRef]
- Walker, J.E.; Luyties, O.; Santangelo, T.J. Factor-dependent archaeal transcription termination. Proc. Natl. Acad. Sci. USA 2017, 114, E6767–E6773. [Google Scholar] [CrossRef]
- Santangelo, T.J.; Cubonova, L.; Skinner, K.M.; Reeve, J.N. Archaeal intrinsic transcription termination in vivo. J. Bacteriol. 2009, 191, 7102–7108. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. The healthy human virome: From virus-host symbiosis to disease. Curr. Opin. Virol. 2021, 47, 86–94. [Google Scholar] [CrossRef]
- Forterre, P.; Prangishvili, D. The major role of viruses in cellular evolution: Facts and hypotheses. Curr. Opin. Virol. 2013, 3, 558–565. [Google Scholar] [CrossRef]
- Tenthorey, J.L.; Emerman, M.; Malik, H.S. Evolutionary Landscapes of Host-Virus Arms Races. Annu. Rev. Immunol. 2022, 40, 271–294. [Google Scholar] [CrossRef] [PubMed]
- Fraser, K.A.; Rice, S.A. Herpes simplex virus immediate-early protein ICP22 triggers loss of serine 2-phosphorylated RNA polymerase II. J. Virol. 2007, 81, 5091–5101. [Google Scholar] [CrossRef] [PubMed]
- Camara, B.; Liu, M.; Reynolds, J.; Shadrin, A.; Liu, B.; Kwok, K.; Simpson, P.; Weinzierl, R.; Severinov, K.; Cota, E.; et al. T7 phage protein Gp2 inhibits the Escherichia coli RNA polymerase by antagonizing stable DNA strand separation near the transcription start site. Proc. Natl. Acad. Sci. USA 2010, 107, 2247–2252. [Google Scholar] [CrossRef]
- Lennon, J.T.; Jones, S.E. Microbial seed banks: The ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 2011, 9, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Summers, W.C. Regulation of RNA metabolism of T7 and related phages. Annu. Rev. Genet. 1972, 6, 191–202. [Google Scholar] [CrossRef]
- Tabib-Salazar, A.; Liu, B.; Shadrin, A.; Burchell, L.; Wang, Z.; Wang, Z.; Goren, M.G.; Yosef, I.; Qimron, U.; Severinov, K.; et al. Full shut-off of Escherichia coli RNA-polymerase by T7 phage requires a small phage-encoded DNA-binding protein. Nucleic Acids Res. 2017, 45, 7697–7707. [Google Scholar] [CrossRef]
- Sokolova, M.L.; Misovetc, I.; Severinov, K.V. Multisubunit RNA Polymerases of Jumbo Bacteriophages. Viruses 2020, 12, 1064. [Google Scholar] [CrossRef]
- Bae, B.; Davis, E.; Brown, D.; Campbell, E.A.; Wigneshweraraj, S.; Darst, S.A. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of sigma70 domain 1.1. Proc. Natl. Acad. Sci. USA 2013, 110, 19772–19777. [Google Scholar] [CrossRef]
- Thomas, D.; Blakqori, G.; Wagner, V.; Banholzer, M.; Kessler, N.; Elliott, R.M.; Haller, O.; Weber, F. Inhibition of RNA polymerase II phosphorylation by a viral interferon antagonist. J. Biol. Chem. 2004, 279, 31471–31477. [Google Scholar] [CrossRef]
- Akhrymuk, I.; Kulemzin, S.V.; Frolova, E.I. Evasion of the innate immune response: The Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J. Virol. 2012, 86, 7180–7191. [Google Scholar] [CrossRef] [Green Version]
- Baquero, D.P.; Contursi, P.; Piochi, M.; Bartolucci, S.; Liu, Y.; Cvirkaite-Krupovic, V.; Prangishvili, D.; Krupovic, M. New virus isolates from Italian hydrothermal environments underscore the biogeographic pattern in archaeal virus communities. ISME J. 2020, 14, 1821–1833. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Kim, S.S.; Lee, J.; Starodubtseva, M.; Cava, F.; Berenguer, J.; Kong, X.P.; Borukhov, S. pH-dependent conformational switch activates the inhibitor of transcription elongation. EMBO J. 2006, 25, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Tagami, S.; Sekine, S.; Kumarevel, T.; Hino, N.; Murayama, Y.; Kamegamori, S.; Yamamoto, M.; Sakamoto, K.; Yokoyama, S. Crystal structure of bacterial RNA polymerase bound with a transcription inhibitor protein. Nature 2010, 468, 978–982. [Google Scholar] [CrossRef]
- Furman, R.; Danhart, E.M.; NandyMazumdar, M.; Yuan, C.; Foster, M.P.; Artsimovitch, I. pH dependence of the stress regulator DksA. PLoS ONE 2015, 10, e0120746. [Google Scholar] [CrossRef] [PubMed]
- Lennon, C.W.; Ross, W.; Martin-Tumasz, S.; Toulokhonov, I.; Vrentas, C.E.; Rutherford, S.T.; Lee, J.H.; Butcher, S.E.; Gourse, R.L. Direct interactions between the coiled-coil tip of DksA and the trigger loop of RNA polymerase mediate transcriptional regulation. Genes Dev. 2012, 26, 2634–2646. [Google Scholar] [CrossRef]
- Kouzine, F.; Wojtowicz, D.; Yamane, A.; Resch, W.; Kieffer-Kwon, K.R.; Bandle, R.; Nelson, S.; Nakahashi, H.; Awasthi, P.; Feigenbaum, L.; et al. Global regulation of promoter melting in naive lymphocytes. Cell 2013, 153, 988–999. [Google Scholar] [CrossRef]
- Pagano, J.M.; Kwak, H.; Waters, C.T.; Sprouse, R.O.; White, B.S.; Ozer, A.; Szeto, K.; Shalloway, D.; Craighead, H.G.; Lis, J.T. Defining NELF-E RNA binding in HIV-1 and promoter-proximal pause regions. PLoS Genet. 2014, 10, e1004090. [Google Scholar] [CrossRef]
- Vos, S.M.; Farnung, L.; Urlaub, H.; Cramer, P. Structure of paused transcription complex Pol II-DSIF-NELF. Nature 2018, 560, 601–606. [Google Scholar] [CrossRef]
- Vorlander, M.K.; Baudin, F.; Moir, R.D.; Wetzel, R.; Hagen, W.J.H.; Willis, I.M.; Muller, C.W. Structural basis for RNA polymerase III transcription repression by Maf1. Nat. Struct. Mol. Biol. 2020, 27, 229–232. [Google Scholar] [CrossRef]
- Zhang, S.; Li, X.; Wang, H.Y.; Steven Zheng, X.F. Beyond regulation of pol III: Role of MAF1 in growth, metabolism, aging and cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 338–343. [Google Scholar] [CrossRef]
- Kim, J.; Cho, J.Y.; Kim, J.W.; Kim, D.G.; Nam, B.H.; Kim, B.S.; Kim, W.J.; Kim, Y.O.; Cheong, J.; Kong, H.J. Molecular Characterization of Paralichthys olivaceus MAF1 and Its Potential Role as an Anti-Viral Hemorrhagic Septicaemia Virus Factor in Hirame Natural Embryo Cells. Int. J. Mol. Sci. 2021, 22, 1353. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Saha, B.; Riley, J.L. The battle over mTOR: An emerging theatre in host-pathogen immunity. PLoS Pathog. 2012, 8, e1002894. [Google Scholar] [CrossRef] [PubMed]
- Prangishvili, D.; Bamford, D.H.; Forterre, P.; Iranzo, J.; Koonin, E.V.; Krupovic, M. The enigmatic archaeal virosphere. Nat. Rev. Microbiol. 2017, 15, 724–739. [Google Scholar] [CrossRef]
- Sheppard, C.; Blombach, F.; Belsom, A.; Schulz, S.; Daviter, T.; Smollett, K.; Mahieu, E.; Erdmann, S.; Tinnefeld, P.; Garrett, R.; et al. Repression of RNA polymerase by the archaeo-viral regulator ORF145/RIP. Nat. Commun. 2016, 7, 13595. [Google Scholar] [CrossRef]
- Sheppard, C.; Werner, F. Structure and mechanisms of viral transcription factors in archaea. Extremophiles 2017, 21, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Pilotto, S.; Fouqueau, T.; Lukoyanova, N.; Sheppard, C.; Lucas-Staat, S.; Diaz-Santin, L.M.; Matelska, D.; Prangishvili, D.; Cheung, A.C.M.; Werner, F. Structural basis of RNA polymerase inhibition by viral and host factors. Nat. Commun. 2021, 12, 5523. [Google Scholar] [CrossRef]
- Prangishvili, D.; Vestergaard, G.; Haring, M.; Aramayo, R.; Basta, T.; Rachel, R.; Garrett, R.A. Structural and genomic properties of the hyperthermophilic archaeal virus ATV with an extracellular stage of the reproductive cycle. J. Mol. Biol. 2006, 359, 1203–1216. [Google Scholar] [CrossRef]
- Dienemann, C.; Schwalb, B.; Schilbach, S.; Cramer, P. Promoter Distortion and Opening in the RNA Polymerase II Cleft. Mol. Cell 2019, 73, 97–106.e104. [Google Scholar] [CrossRef]
- Grohmann, D.; Nagy, J.; Chakraborty, A.; Klose, D.; Fielden, D.; Ebright, R.H.; Michaelis, J.; Werner, F. The initiation factor TFE and the elongation factor Spt4/5 compete for the RNAP clamp during transcription initiation and elongation. Mol. Cell 2011, 43, 263–274. [Google Scholar] [CrossRef]
- Schulz, S.; Gietl, A.; Smollett, K.; Tinnefeld, P.; Werner, F.; Grohmann, D. TFE and Spt4/5 open and close the RNA polymerase clamp during the transcription cycle. Proc. Natl. Acad. Sci. USA 2016, 113, E1816–E1825. [Google Scholar] [CrossRef] [Green Version]
- Hausner, W.; Lange, U.; Musfeldt, M. Transcription factor S, a cleavage induction factor of the archaeal RNA polymerase. J. Biol. Chem. 2000, 275, 12393–12399. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.C.; Cramer, P. Structural basis of RNA polymerase II backtracking, arrest and reactivation. Nature 2011, 471, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Lee, J.; Lomakin, I.; Borukhov, S. Transcript cleavage factors GreA and GreB act as transient catalytic components of RNA polymerase. EMBO J. 2003, 22, 6322–6334. [Google Scholar] [CrossRef] [PubMed]
- Roghanian, M.; Yuzenkova, Y.; Zenkin, N. Controlled interplay between trigger loop and Gre factor in the RNA polymerase active centre. Nucleic Acids Res. 2011, 39, 4352–4359. [Google Scholar] [CrossRef]
- Vassylyeva, M.N.; Svetlov, V.; Dearborn, A.D.; Klyuyev, S.; Artsimovitch, I.; Vassylyev, D.G. The carboxy-terminal coiled-coil of the RNA polymerase beta’-subunit is the main binding site for Gre factors. EMBO Rep. 2007, 8, 1038–1043. [Google Scholar] [CrossRef]
- Engel, C.; Sainsbury, S.; Cheung, A.C.; Kostrewa, D.; Cramer, P. RNA polymerase I structure and transcription regulation. Nature 2013, 502, 650–655. [Google Scholar] [CrossRef]
- Neyer, S.; Kunz, M.; Geiss, C.; Hantsche, M.; Hodirnau, V.V.; Seybert, A.; Engel, C.; Scheffer, M.P.; Cramer, P.; Frangakis, A.S. Structure of RNA polymerase I transcribing ribosomal DNA genes. Nature 2016, 540, 607–610. [Google Scholar] [CrossRef]
- Girbig, M.; Misiaszek, A.D.; Vorlander, M.K.; Lafita, A.; Grotsch, H.; Baudin, F.; Bateman, A.; Muller, C.W. Cryo-EM structures of human RNA polymerase III in its unbound and transcribing states. Nat. Struct. Mol. Biol. 2021, 28, 210–219. [Google Scholar] [CrossRef]
- Li, S.; Ding, B.; Chen, R.; Ruggiero, C.; Chen, X. Evidence that the transcription elongation function of Rpb9 is involved in transcription-coupled DNA repair in Saccharomyces cerevisiae. Mol. Cell Biol. 2006, 26, 9430–9441. [Google Scholar] [CrossRef]
- Lange, U.; Hausner, W. Transcriptional fidelity and proofreading in Archaea and implications for the mechanism of TFS-induced RNA cleavage. Mol. Microbiol. 2004, 52, 1133–1143. [Google Scholar] [CrossRef]
- Prescott, E.M.; Osheim, Y.N.; Jones, H.S.; Alen, C.M.; Roan, J.G.; Reeder, R.H.; Beyer, A.L.; Proudfoot, N.J. Transcriptional termination by RNA polymerase I requires the small subunit Rpa12p. Proc. Natl. Acad. Sci. USA 2004, 101, 6068–6073. [Google Scholar] [CrossRef] [PubMed]
- Landrieux, E.; Alic, N.; Ducrot, C.; Acker, J.; Riva, M.; Carles, C. A subcomplex of RNA polymerase III subunits involved in transcription termination and reinitiation. EMBO J. 2006, 25, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Balaban, N.Q.; Baquero, F.; Courvalin, P.; Glaser, P.; Gophna, U.; Kishony, R.; Molin, S.; Tonjum, T. Antibiotic resistance: Turning evolutionary principles into clinical reality. FEMS Microbiol. Rev. 2020, 44, 171–188. [Google Scholar] [CrossRef]
- Aristoff, P.A.; Garcia, G.A.; Kirchhoff, P.D.; Showalter, H.D. Rifamycins—Obstacles and opportunities. Tuberculosis 2010, 90, 94–118. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef]
- Zisi, A.; Bartek, J.; Lindstrom, M.S. Targeting Ribosome Biogenesis in Cancer: Lessons Learned and Way Forward. Cancers 2022, 14, 2126. [Google Scholar] [CrossRef]
- Ferreira, R.; Schneekloth, J.S., Jr.; Panov, K.I.; Hannan, K.M.; Hannan, R.D. Targeting the RNA Polymerase I Transcription for Cancer Therapy Comes of Age. Cells 2020, 9, 266. [Google Scholar] [CrossRef]
- Martin, R.D.; Hebert, T.E.; Tanny, J.C. Therapeutic Targeting of the General RNA Polymerase II Transcription Machinery. Int. J. Mol. Sci. 2020, 21, 3354. [Google Scholar] [CrossRef]
- Laptenko, O.; Prives, C. Transcriptional regulation by p53: One protein, many possibilities. Cell Death Differ. 2006, 13, 951–961. [Google Scholar] [CrossRef]
- Poortinga, G.; Quinn, L.M.; Hannan, R.D. Targeting RNA polymerase I to treat MYC-driven cancer. Oncogene 2015, 34, 403–412. [Google Scholar] [CrossRef]
- Mosaei, H.; Harbottle, J. Mechanisms of antibiotics inhibiting bacterial RNA polymerase. Biochem. Soc. Trans. 2019, 47, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, S.H.; Haeckl, F.P.J.; Muller, R. Beyond the approved: Target sites and inhibitors of bacterial RNA polymerase from bacteria and fungi. Nat. Prod. Rep. 2022, 39, 1226–1263. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.X.; Hudson, B.P.; Das, K.; Arnold, E.; Ebright, R.H. Structures of RNA polymerase-antibiotic complexes. Curr. Opin. Struct. Biol. 2009, 19, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Mandal, S.; Degen, D.; Liu, Y.; Ebright, Y.W.; Li, S.; Feng, Y.; Zhang, Y.; Mandal, S.; Jiang, Y.; et al. Structural Basis of Mycobacterium tuberculosis Transcription and Transcription Inhibition. Mol. Cell 2017, 66, 169–179.e168. [Google Scholar] [CrossRef] [PubMed]
- Temiakov, D.; Zenkin, N.; Vassylyeva, M.N.; Perederina, A.; Tahirov, T.H.; Kashkina, E.; Savkina, M.; Zorov, S.; Nikiforov, V.; Igarashi, N.; et al. Structural basis of transcription inhibition by antibiotic streptolydigin. Mol. Cell 2005, 19, 655–666. [Google Scholar] [CrossRef]
- Molodtsov, V.; Fleming, P.R.; Eyermann, C.J.; Ferguson, A.D.; Foulk, M.A.; McKinney, D.C.; Masse, C.E.; Buurman, E.T.; Murakami, K.S. X-ray crystal structures of Escherichia coli RNA polymerase with switch region binding inhibitors enable rational design of squaramides with an improved fraction unbound to human plasma protein. J. Med. Chem. 2015, 58, 3156–3171. [Google Scholar] [CrossRef]
- Lin, W.; Das, K.; Degen, D.; Mazumder, A.; Duchi, D.; Wang, D.; Ebright, Y.W.; Ebright, R.Y.; Sineva, E.; Gigliotti, M.; et al. Structural Basis of Transcription Inhibition by Fidaxomicin (Lipiarmycin A3). Mol. Cell 2018, 70, 60–71.e15. [Google Scholar] [CrossRef]
- Braffman, N.R.; Piscotta, F.J.; Hauver, J.; Campbell, E.A.; Link, A.J.; Darst, S.A. Structural mechanism of transcription inhibition by lasso peptides microcin J25 and capistruin. Proc. Natl. Acad. Sci. USA 2019, 116, 1273–1278. [Google Scholar] [CrossRef]
- Degen, D.; Feng, Y.; Zhang, Y.; Ebright, K.Y.; Ebright, Y.W.; Gigliotti, M.; Vahedian-Movahed, H.; Mandal, S.; Talaue, M.; Connell, N.; et al. Transcription inhibition by the depsipeptide antibiotic salinamide A. eLife 2014, 3, e02451. [Google Scholar] [CrossRef]
- Kaplan, C.D.; Larsson, K.M.; Kornberg, R.D. The RNA polymerase II trigger loop functions in substrate selection and is directly targeted by alpha-amanitin. Mol. Cell 2008, 30, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Vassylyev, D.G.; Vassylyeva, M.N.; Perederina, A.; Tahirov, T.H.; Artsimovitch, I. Structural basis for transcription elongation by bacterial RNA polymerase. Nature 2007, 448, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Vassylyev, D.G.; Vassylyeva, M.N.; Zhang, J.; Palangat, M.; Artsimovitch, I.; Landick, R. Structural basis for substrate loading in bacterial RNA polymerase. Nature 2007, 448, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Wang, D.; Ebright, Y.W.; Korlann, Y.; Kortkhonjia, E.; Kim, T.; Chowdhury, S.; Wigneshweraraj, S.; Irschik, H.; Jansen, R.; et al. Opening and closing of the bacterial RNA polymerase clamp. Science 2012, 337, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, F.; Cramer, P. Structural basis of transcription inhibition by alpha-amanitin and implications for RNA polymerase II translocation. Nat. Struct. Mol. Biol. 2008, 15, 811–818. [Google Scholar] [CrossRef]
- Liu, X.; Farnung, L.; Wigge, C.; Cramer, P. Cryo-EM structure of a mammalian RNA polymerase II elongation complex inhibited by alpha-amanitin. J. Biol. Chem. 2018, 293, 7189–7194. [Google Scholar] [CrossRef]
- Mukhopadhyay, J.; Das, K.; Ismail, S.; Koppstein, D.; Jang, M.; Hudson, B.; Sarafianos, S.; Tuske, S.; Patel, J.; Jansen, R.; et al. The RNA polymerase “switch region” is a target for inhibitors. Cell 2008, 135, 295–307. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilotto, S.; Werner, F. How to Shut Down Transcription in Archaea during Virus Infection. Microorganisms 2022, 10, 1824. https://doi.org/10.3390/microorganisms10091824
Pilotto S, Werner F. How to Shut Down Transcription in Archaea during Virus Infection. Microorganisms. 2022; 10(9):1824. https://doi.org/10.3390/microorganisms10091824
Chicago/Turabian StylePilotto, Simona, and Finn Werner. 2022. "How to Shut Down Transcription in Archaea during Virus Infection" Microorganisms 10, no. 9: 1824. https://doi.org/10.3390/microorganisms10091824