
Immunoinformatic Execution and Design of an Anti-Epstein–Barr Virus Vaccine with Multiple Epitopes Triggering Innate and Adaptive Immune Responses
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Conservancy Analysis of the Target Protein
2.2. Characterization of Potential Epitopes
2.2.1. B-Lymphocytes-Specific Epitopes
2.2.2. CTL-Specific Epitopes
2.2.3. HTL-Specific Epitopes
2.3. Immunogenicity of the Selected Epitopes
2.4. Evaluation of the Population Coverage of the Finalized Epitopes
2.5. Final Vaccine Construct
2.6. Validation of the Immunogenicity of the Construct
2.7. Modeling the Construct
2.8. Physicochemical Properties and Structural Validation of the Construct
2.9. Prediction of Immune Stimulation
2.10. Prediction of the Discontinuous Epitopes of the Final Construct
2.11. Binding Pocket and Molecular Interaction Analysis of the Construct
2.12. Molecular Dynamics Simulation
2.13. Codon Optimization and Expression Analysis
3. Results
3.1. Conservancy Analysis of the Target Protein
3.2. Characterization of Potential Epitopes
3.2.1. B-Lymphocytes-Specific Epitopes
3.2.2. CTL-Specific Epitopes
3.2.3. HTL-Specific Epitopes
3.3. Evaluation of the Population Coverage of the Finalized Epitopes
3.4. Final Vaccine Construct
3.5. Validation of the Immunogenicity of the Construct
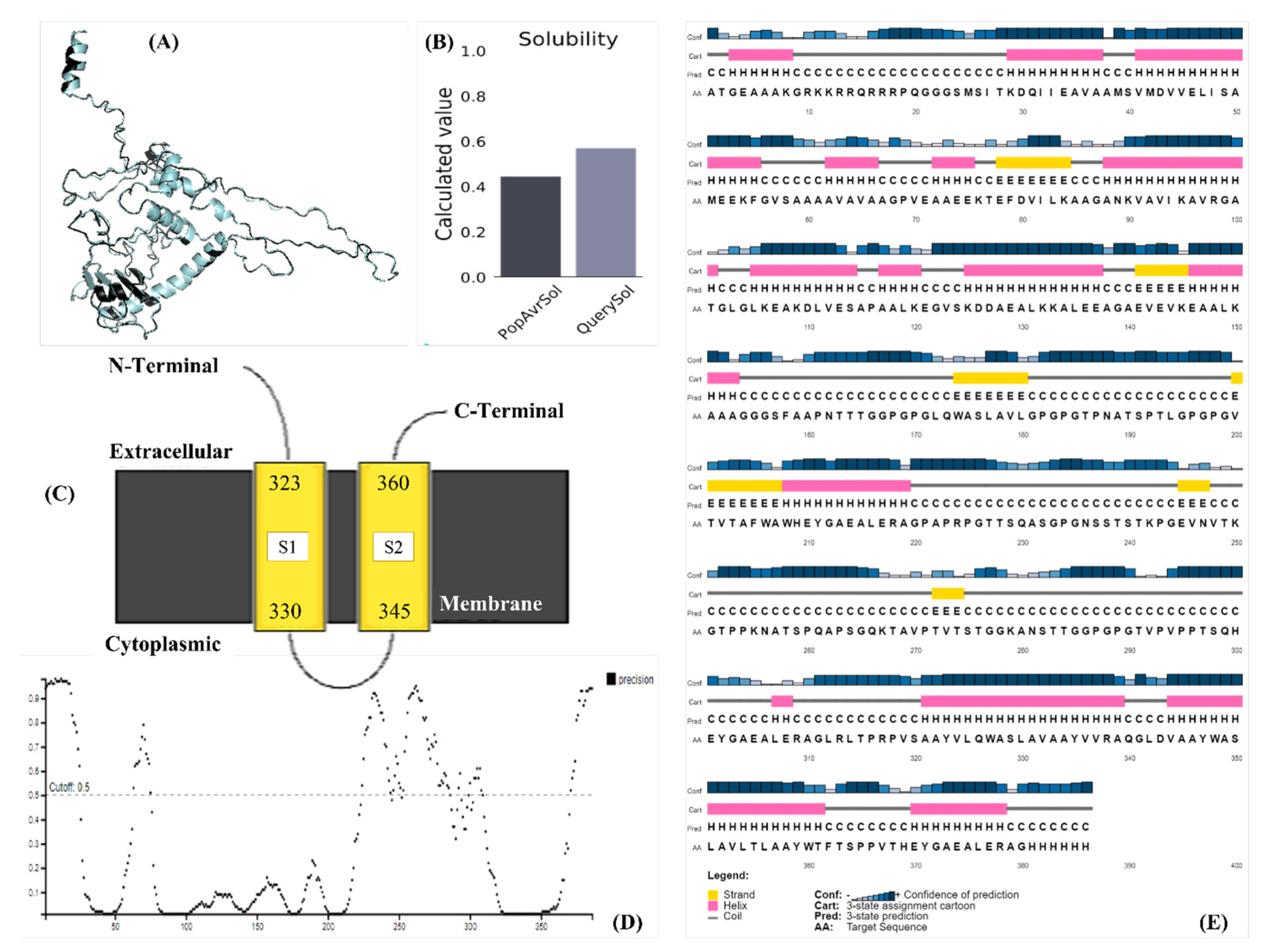
3.6. Modeling the Construct
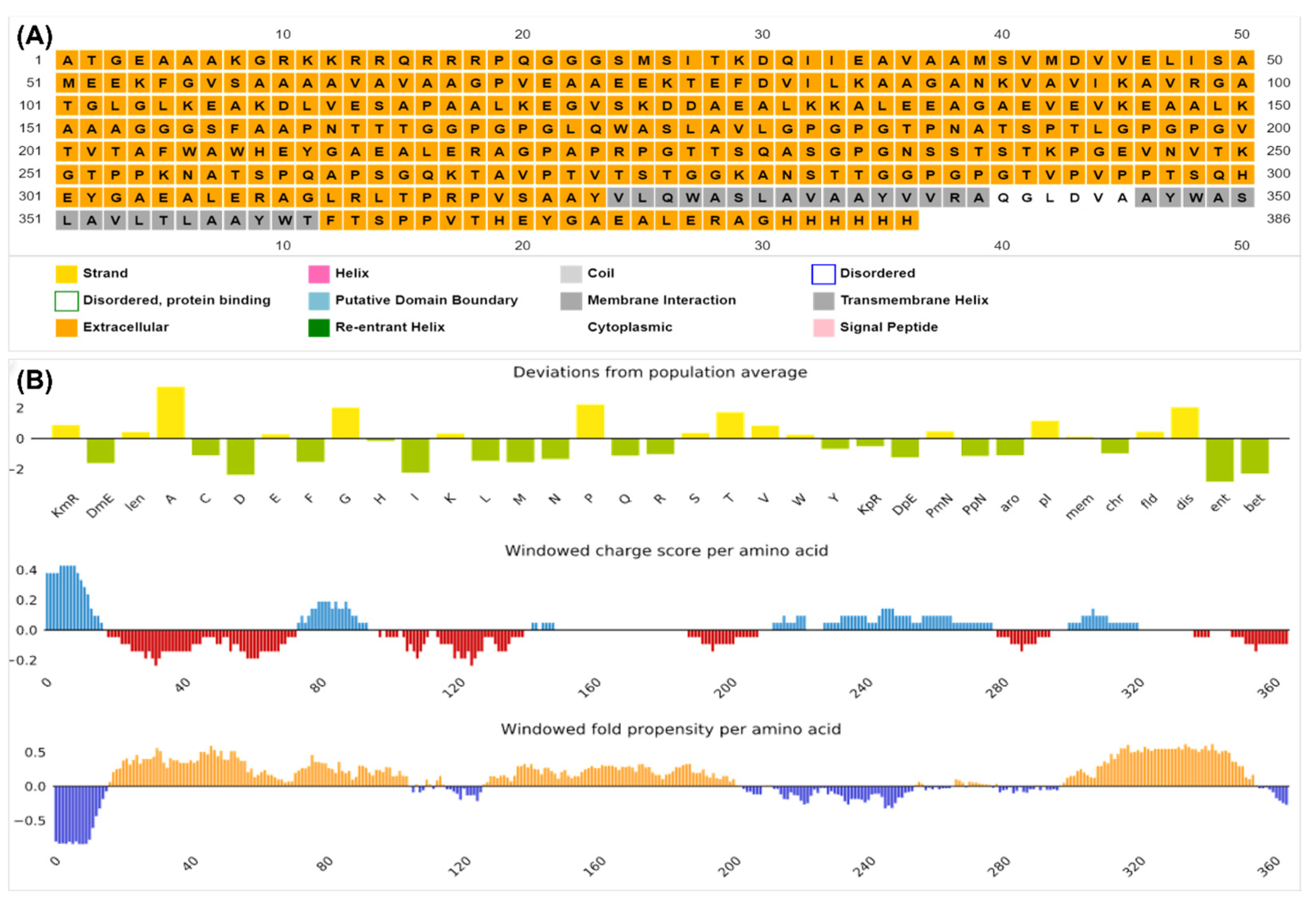
3.7. Physicochemical Properties and Structural Validation of the Construct
3.8. Prediction of Immune Stimulation
3.9. Prediction of the Discontinuous Epitopes of the Final Construct
3.10. Binding Pocket and Molecular Interaction Analysis of the Construct
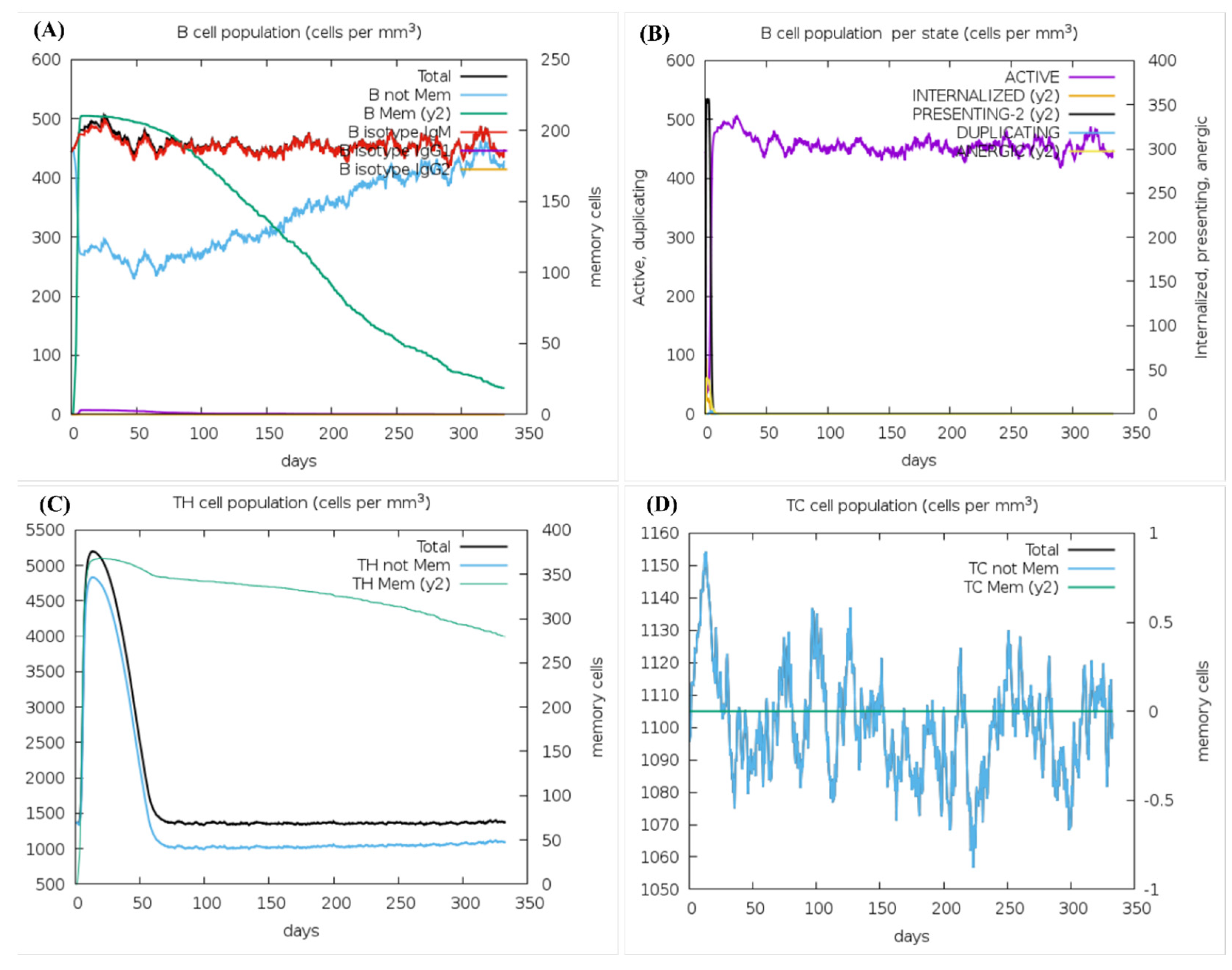
3.11. Molecular Dynamics Simulation
3.12. Codon Optimization and Expression Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vockerodt, M.; Yap, L.F.; Shannon-Lowe, C.; Curley, H.; Wei, W.; Vrzalikova, K.; Murray, P.G. The Epstein–Barr virus and the pathogenesis of lymphoma. J. Pathol. 2015, 235, 312–322. [Google Scholar] [CrossRef]
- Lee, S.; Sakakibara, S.; Maruo, S.; Zhao, B.; Calderwood, M.A.; Holthaus, A.M.; Lai, C.-Y.; Takada, K.; Kieff, E.; Johannsen, E. Epstein-Barr virus nuclear protein 3C domains necessary for lymphoblastoid cell growth: Interaction with RBP-Jκ regulates TCL1. J. Virol. 2009, 83, 12368–12377. [Google Scholar] [CrossRef]
- Gottschalk, S.; Ng, C.Y.; Perez, M.; Smith, C.A.; Sample, C.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. An Epstein-Barr virus deletion mutant associated with fatal lymphoproliferative disease unresponsive to therapy with virus-specific CTLs. Blood J. Am. Soc. Hematol. 2001, 97, 835–843. [Google Scholar] [CrossRef]
- Macedo, C.; Webber, S.A.; Donnenberg, A.D.; Popescu, I.; Hua, Y.; Green, M.; Rowe, D.; Smith, L.; Brooks, M.M.; Metes, D. EBV-specific CD8+ T cells from asymptomatic pediatric thoracic transplant patients carrying chronic high EBV loads display contrasting features: Activated phenotype and exhausted function. J. Immunol. 2011, 186, 5854–5862. [Google Scholar] [CrossRef]
- Rostgaard, K.; Balfour Jr, H.H.; Jarrett, R.; Erikstrup, C.; Pedersen, O.; Ullum, H.; Nielsen, L.P.; Voldstedlund, M.; Hjalgrim, H. Primary Epstein-Barr virus infection with and without infectious mononucleosis. PLoS ONE 2019, 14, e0226436. [Google Scholar] [CrossRef]
- Hutt-Fletcher, L. Epstein–Barr Virus. In Cancers in People with HIV and AIDS; Springer: Berlin/Heidelberg, Germany, 2014; pp. 75–85. [Google Scholar]
- Münz, C. Epstein–Barr virus-specific immune control by innate lymphocytes. Front. Immunol. 2017, 8, 1658. [Google Scholar] [CrossRef]
- Pattle, S.B.; Farrell, P.J. The role of Epstein–Barr virus in cancer. Expert Opin. Biol. Ther. 2006, 6, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Co-Stimulatory Molecules during Immune Control of Epstein Barr Virus Infection. Biomolecules 2021, 12, 38. [Google Scholar] [CrossRef] [PubMed]
- Naveed, M.; Ali, U.; Karobari, M.I.; Ahmed, N.; Mohamed, R.N.; Abullais, S.S.; Kader, M.A.; Marya, A.; Messina, P.; Scardina, G.A. A Vaccine Construction against COVID-19-Associated Mucormycosis Contrived with Immunoinformatics-Based Scavenging of Potential Mucoralean Epitopes. Vaccines 2022, 10, 664. [Google Scholar] [CrossRef] [PubMed]
- Naveed, M.; Hassan, J.-U.; Ahmad, M.; Naeem, N.; Mughal, M.S.; Rabaan, A.A.; Aljeldah, M.; Shammari, B.R.A.; Alissa, M.; Sabour, A.A. Designing mRNA-and Peptide-Based Vaccine Construct against Emerging Multidrug-Resistant Citrobacter freundii: A Computational-Based Subtractive Proteomics Approach. Medicina 2022, 58, 1356. [Google Scholar] [CrossRef] [PubMed]
- Naveed, M.; Yaseen, A.R.; Khalid, H.; Ali, U.; Rabaan, A.A.; Garout, M.; Halwani, M.A.; Al Mutair, A.; Alhumaid, S.; Al Alawi, Z. Execution and Design of an Anti HPIV-1 Vaccine with Multiple Epitopes Triggering Innate and Adaptive Immune Responses: An Immunoinformatic Approach. Vaccines 2022, 10, 869. [Google Scholar] [CrossRef] [PubMed]
- Tomar, N. Immunoinformatics; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Naveed, M.; Bukhari, B.; Afzal, N.; Sadia, H.; Meer, B.; Riaz, T.; Ali, U.; Ahmed, N. Geographical, Molecular, and Computational Analysis of Migraine-Causing Genes. J. Comput. Biophys. Chem. 2021, 20, 391–403. [Google Scholar] [CrossRef]
- Emini, E.A.; Hughes, J.V.; Perlow, D.; Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839. [Google Scholar] [CrossRef]
- Karplus, P.; Schulz, G. Prediction of chain flexibility in proteins: A tool for the selection of peptide antigens. Naturwissenschaften 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Pellequer, J.-L.; Westhof, E.; Van Regenmortel, M.H. Correlation between the location of antigenic sites and the prediction of turns in proteins. Immunol. Lett. 1993, 36, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Guo, D.; Hodges, R. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: Correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry 1986, 25, 5425–5432. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, J.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 1–8. [Google Scholar] [CrossRef]
- Kringelum, J.V.; Lundegaard, C.; Lund, O.; Nielsen, M. Reliable B cell epitope predictions: Impacts of method development and improved benchmarking. PLoS Comput. Biol. 2012, 8, e1002829. [Google Scholar] [CrossRef]
- Nielsen, M.; Lund, O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinform. 2009, 10, 1–10. [Google Scholar] [CrossRef]
- Bui, H.-H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 1–5. [Google Scholar] [CrossRef]
- Sanches, R.C.; Tiwari, S.; Ferreira, L.C.; Oliveira, F.M.; Lopes, M.D.; Passos, M.J.; Maia, E.H.; Taranto, A.G.; Kato, R.; Azevedo, V.A. Immunoinformatics design of multi-epitope peptide-based vaccine against Schistosoma mansoni using transmembrane proteins as a target. Front. Immunol. 2021, 12, 621706. [Google Scholar] [CrossRef] [PubMed]
- Naveed, M.; Sheraz, M.; Amin, A.; Waseem, M.; Aziz, T.; Khan, A.A.; Ghani, M.; Shahzad, M.; Alruways, M.W.; Dablool, A.S. Designing a novel peptide-based multi-epitope vaccine to evoke a robust immune response against pathogenic multidrug-resistant Providencia heimbachae. Vaccines 2022, 10, 1300. [Google Scholar] [CrossRef] [PubMed]
- Sameer, A.S.; Nissar, S. Toll-like receptors (TLRs): Structure, functions, signaling, and role of their polymorphisms in colorectal cancer susceptibility. BioMed Res. Int. 2021, 2021, 1157023. [Google Scholar] [CrossRef]
- Cohen, J.I. Epstein–barr virus vaccines. Clin. Transl. Immunol. 2015, 4, e32. [Google Scholar] [CrossRef]
- Smith, C.; Lee, V.; Schuessler, A.; Beagley, L.; Rehan, S.; Tsang, J.; Li, V.; Tiu, R.; Smith, D.; Neller, M.A. Pre-emptive and therapeutic adoptive immunotherapy for nasopharyngeal carcinoma: Phenotype and effector function of T cells impact on clinical response. Oncoimmunology 2017, 6, e1273311. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.R.; Wilson, D.F. Evidence of Epstein-Barr virus association with head and neck cancers: A review. J. Can. Dent. Assoc. 2016, 82, 1–11. [Google Scholar]
- Tempera, I.; Lieberman, P.M. Epigenetic regulation of EBV persistence and oncogenesis. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Ciążyńska, M.; Bednarski, I.A.; Wódz, K.; Narbutt, J.; Lesiak, A. NLRP1 and NLRP3 inflammasomes as a new approach to skin carcinogenesis. Oncol. Lett. 2020, 19, 1649–1656. [Google Scholar] [CrossRef]
- Taylor, G.S.; Jia, H.; Harrington, K.; Lee, L.W.; Turner, J.; Ladell, K.; Price, D.A.; Tanday, M.; Matthews, J.; Roberts, C. A recombinant modified vaccinia ankara vaccine encoding Epstein–Barr virus (EBV) target antigens: A phase I trial in UK patients with EBV-positive cancer. Clin. Cancer Res. 2014, 20, 5009–5022. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef]
- Kelly, G.; Bell, A.; Rickinson, A. Epstein–Barr virus–associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat. Med. 2002, 8, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- De Bree, L.; Koeken, V.A.; Joosten, L.A.; Aaby, P.; Benn, C.S.; van Crevel, R.; Netea, M.G. Non-specific effects of vaccines: Current evidence and potential implications. In Seminars in Immunology; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Benn, C.S.; Fisker, A.B.; Rieckmann, A.; Jensen, A.K.G.; Aaby, P. How to evaluate potential non-specific effects of vaccines: The quest for randomized trials or time for triangulation? Expert Rev. Vaccines 2018, 17, 411–420. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epitopes | Antigenicity (0.4) | B-Turn (1.061) | Hydrophilicity (2.382) | Flexibility (1.023) | Accessibility (1.000) |
| B-lymphocytes-specific epitopes | |||||
| PAPRPGTTSQASGPGNSSTSTKPGEVNVTKGTPPKNATSPQAPSGQKTAVPTVTSTGGKANSTTG | 0.984 | 1.702 | 4.7372 | 1.076 | 1.323 |
| TVPVPPTSQ | 1.1033 | 1.1633 | 2.873 | 1.225 | 1.633 |
| Epitopes | Antigenicity | Restricting HLA Alleles | |||
| MHC-I-restricted epitopes | |||||
| FAAPNTTTG | 0.6798 | HLA-B*35:01, HLA-C*03:03, HLA-C*12:03 | |||
| LQWASLAVL | 1.6217 | HLA-A*02:06, HLA-B*15:01, HLA-B*48:01 | |||
| TPNATSPTL | 0.4187 | HLA-B*39:01, HLA-B*35:01 | |||
| VTVTAFWAW | 0.6731 | HLA-B*58:01, HLA-B*57:01 | |||
| MHC-II-restricted epitopes | |||||
| LRLTPRPVS | 2.6164 | HLA-DQA1*02:01/DQB1*04:02, HLA-DPA1*01:03/DPB1*03:01, HLA-DQA1*05:01/DQB1*04:02, HLA-DRB1*11:01, HLA-DRB1*13:01, HLA-DRB3*03:01, HLA-DRB1*08:02, HLA-DRB3*02:02, HLA-DRB1*08:01, HLA-DRB1*03:01, HLA-DQA1*01:02/DQB1*05:01 | |||
| VLQWASLAV | 0.9940 | HLA-DQA1*02:01/DQB1*03:03, HLA-DPA1*03:01/DPB1*04:02, HLA-DRB4*01:01, HLA-DRB1*04:05, HLA-DRB1*15:01, HLA-DRB4*01:03, HLA-DQA1*01:02/DQB1*05:01, HLA-DRB1*13:01, HLA-DPA1*01:03/DPB1*06:01, HLA-DQA1*02:01/DQB1*03:01 | |||
| VVRAQGLDV | 0.8213 | HLA-DRB4*01:01, HLA-DRB1*09:01, HLA-DRB1*07:01, HLA-DRB1*13:02, HLA-DRB3*03:01, HLA-DRB1*01:01, HLA-DRB4*01:03 | |||
| WASLAVLTL | 1.0200 | HLA-DRB1*09:01, HLA-DRB1*10:01, HLA-DRB4*01:03, HLA-DRB1*13:01 | |||
| WIFTSPPVT | 0.4742 | HLA-DQA1*06:01/DQB1*04:02, HLA-DQA1*02:01/DQB1*04:02, HLA-DRB1*07:01, HLA-DQA1*05:01/DQB1*04:02, HLA-DRB1*10:01, HLA-DRB1*01:01, HLA-DQA1*01:02/DQB1*05:01 | |||
| Analysis | Result; Score (Threshold) |
|---|---|
| Antigenicity | Antigenic; 0.5710 (0.5000) |
| Allergenicity | Non-allergen |
| Toxicity | Non-toxic |
| IFN-gamma stimulation for epitope 1 | Positive; 0.318 (0.000) |
| IFN-gamma stimulation for epitope 2 | Positive; 0.161 (0.000) |
| IFN-gamma stimulation for epitope 3 | Positive; 0.555 (0.000) |
| IFN-gamma stimulation for epitope 4 | Positive; 0.162 (0.000) |
| IFN-gamma stimulation for epitope 5 | Negative; −0.8088 (0.000) |
| Non-homology analysis against human proteome | Non-homologous |
| Non-homology analysis against gut microbiota | Non-homologous |
| No. of amino acids | 386 |
| Molecular weight | 38,966.82 |
| Theoretical pI | 8.06 |
| Estimated half-life in mammalian reticulocytes | 4.4 h |
| Instability index | Stable; 33.94 (<34) |
| Aliphatic index | Thermostable; 73.76 |
| GRAVY | Hydrophilic; −0.195 (<0) |
| Solubility upon overexpression (Scratch) | Soluble; 0.955 (0.5) |
| GO Term | Name | Prob |
|---|---|---|
| Biological Process | ||
| GO:0006396 | RNA processing | 0.6 |
| GO:0010468 | regulation of gene expression | 0.6 |
| GO:0000398 | mRNA splicing, via spliceosome | 0.62 |
| GO:0009059 | macromolecule biosynthetic process | 0.63 |
| GO:0006351 | transcription, DNA-templated | 0.65 |
| GO:0006355 | regulation of transcription, DNA-templated | 0.7 |
| GO:0051252 | regulation of RNA metabolic process | 0.71 |
| GO:0006810 | transport | 0.71 |
| GO:0008380 | RNA splicing | 0.73 |
| GO:0051171 | regulation of nitrogen metabolic process | 0.73 |
| GO:0034645 | cellular macromolecule biosynthetic process | 0.8 |
| GO:2001141 | regulation of RNA biosynthetic process | 0.8 |
| GO:0019222 | regulation of metabolic process | 0.8 |
| GO:1903506 | regulation of nucleic acid-templated transcription | 0.82 |
| Molecular Functions | ||
| GO:0003676 | nucleic acid binding | 0.97 |
| GO:0044822 | poly(A) RNA binding | 0.87 |
| GO:0008092 | cytoskeletal protein binding | 0.82 |
| GO:0003723 | RNA binding | 0.8 |
| GO:0000166 | nucleotide binding | 0.79 |
| GO:0019900 | kinase binding | 0.74 |
| GO:0003824 | catalytic activity | 0.65 |
| GO:0003779 | actin binding | 0.64 |
| GO:0015631 | tubulin binding | 0.63 |
| GO:0003677 | DNA binding | 0.63 |
| GO:0008017 | microtubule binding | 0.6 |
| GO:0032549 | ribonucleoside binding | 0.59 |
| GO:0001664 | G-protein coupled receptor binding | 0.59 |
| GO:0001883 | purine nucleoside binding | 0.58 |
| GO:0017076 | purine nucleotide binding | 0.56 |
| GO:0035639 | purine ribonucleoside triphosphate binding | 0.55 |
| GO:0016817 | hydrolase activity, acting on acid anhydrides | 0.52 |
| Cellular Functions | ||
| GO:0005739 | mitochondrion | 0.83 |
| GO:0031224 | intrinsic component of membrane | 0.71 |
| GO:0016020 | membrane | 0.7 |
| GO:0031966 | mitochondrial membrane | 0.61 |
| GO:0005886 | plasma membrane | 0.58 |
| GO:0016021 | integral component of membrane | 0.53 |
| GO:0030529 | ribonucleoprotein complex | 0.53 |
| Score | Coord_x | Coord_y | Coord_z | Residues |
|---|---|---|---|---|
| 0.44332 | 101.61 | 93.8105 | 84.3158 | A_43_ASP; A_47_LEU |
| 0.3123 | 74.7601 | 89.5706 | 139.954 | A_372_GLY; A_375_ALA; A_376_LEU; A_379_ALA; A_380_GLY |
| 0.30649 | 98.8762 | 95.9583 | 81.1978 | A_48_ILE; A_51_MET |
| 0.27802 | 103.259 | 96.5484 | 87.0455 | A_141_GLU; A_178_ALA; A_179_VAL; A_180_LEU |
| 0.24589 | 69.878 | 91.1676 | 135.446 | A_369_HIS; A_374_GLU; A_375_ALA; A_378_ARG; A_379_ALA |
| 0.24362 | 75.9403 | 88.6986 | 105.536 | A_98_ARG; A_102_GLY; A_103_LEU; A_104_GLY; A_105_LEU; A_314_LEU; A_315_THR; A_316_PRO; A_317_ARG |
| 0.2169 | 97.332 | 93.4671 | 84.5491 | A_51_MET; A_137_GLU; A_139_GLY; A_141_GLU |
| 0.21223 | 101.958 | 100.489 | 85.6208 | A_179_VAL; A_180_LEU; A_181_GLY; A_206_TRP |
| 0.19845 | 105.508 | 92.7975 | 87.0885 | A_8_LYS; A_43_ASP; A_170_PRO; A_174_TRP; A_178_ALA |
| 0.15525 | 72.1554 | 84.3839 | 97.2013 | A_105_LEU; A_310_ALA; A_311_GLY; A_312_LEU; A_313_ARG |
| 0.14171 | 74.9699 | 89.9079 | 109.34 | A_103_LEU; A_104_GLY; A_316_PRO; A_317_ARG |
| 0.14057 | 104.133 | 97.4513 | 82.7488 | A_43_ASP; A_180_LEU |
| 0.13555 | 111.707 | 102.763 | 81.6146 | A_36_VAL; A_208_TRP; A_230_GLN |
| 0.1255 | 74.1947 | 82.6675 | 101.329 | A_98_ARG; A_105_LEU; A_311_GLY; A_312_LEU; A_313_ARG |
| 0.12307 | 69.1664 | 88.2258 | 141.794 | A_378_ARG; A_379_ALA; A_380_GLY; A_381_HIS; A_382_HIS; A_383_HIS; A_384_HIS |
| 0.12226 | 106.456 | 78.4865 | 93.9053 | A_9_GLY; A_10_ARG; A_13_ARG; A_255_LYS |
| 0.11209 | 91.481 | 91.5907 | 100.331 | A_127_ASP |
| 0.10931 | 109.269 | 99.044 | 83.7031 | A_36_VAL; A_40_SER; A_178_ALA; A_180_LEU; A_208_TRP; A_210_GLU |
| 0.1084 | 106.421 | 101.195 | 80.0099 | A_180_LEU; A_208_TRP |
| 0.10469 | 75.056 | 82.1306 | 110.437 | A_349_ALA; A_350_SER; A_351_LEU |
| Interacting Residues | ||||
|---|---|---|---|---|
| Sr. | Vaccine AA Residue | Receptor AA Residue | Bond Length (Angstrom) | Bond Type |
| 1 | His385 | Thr391 | 3.0 | Amine (hydrogen) |
| 2 | His381 | Asp419 | 1.9 | Conventional covalent bond |
| 3 | Leu105 | Asp31 | 2.4 | H-bond–van der Waals transition |
| 4 | Glu107 | Cys30 | 2.8 | Hydrogen |
| 5 | Glu377 | Lys422 | 1.8 | Conventional covalent bond |
| 6 | Glu377 | Lys422 | 1.8 | Conventional covalent bond |
| 7 | Lys120 | Glu526 | 1.7 | Conventional covalent bond |
| 8 | Ser157 | Ser524 | 1.9 | Conventional covalent bond |
| 9 | Ala321 | Lys527 | 1.7 | Conventional covalent bond |
| 10 | Trp327 | Glu481 | 2.5 | H-bond–van der Waals transition |
| 11 | Trp327 | Glu481 | 2.0 | H-bond–van der Waals transition |
| 12 | Tyr371 | Arg486 | 2.0 | H-bond–van der Waals transition |
| 13 | Tyr371 | Arg486 | 2.7 | Hydroxyl (hydrogen) |
| 14 | Gln326 | Asp419 | 2.0 | H-bond–van der Waals transition |
| 15 | Gln326 | Typ440 | 2.7 | Hydroxyl (hydrogen) |
| 16 | Ser326 | Arg340 | 1.9 | Conventional covalent bond |
| 17 | Leu330 | Arg340 | 2.4 | H-bond–van der Waals transition |
| 18 | Lys242 | Glu310 | 1.8 | Conventional covalent bond |
| 19 | Lys242 | Glu310 | 1.9 | Conventional covalent bond |
| 20 | Pro221 | Lys308 | 1.7 | Conventional covalent bond |
| 21 | Arg10 | Asn199 | 2.6 | Carboxylic (hydrogen) |
| 22 | Arg10 | Asn199 | 1.8 | Conventional covalent bond |
| 23 | Glu173 | Glu225 | 2.7 | Hydroxyl (hydrogen) |
| 24 | Arg338 | Glu152 | 2.5 | H-bond–van der Waals transition |
| 25 | Arg338 | Glu152 | 1.8 | Conventional covalent bond |
| 26 | Ala345 | Glu103 | 2.9 | Oxazole (hydrogen) |
| 27 | Tyr335 | Glu177 | 1.9 | Conventional covalent bond |
| 28 | Arg338 | Glu177 | 1.8 | Conventional covalent bond |
| 29 | Tyr371 | Arg508 | 1.9 | Conventional covalent bond |
| 30 | Tyr371 | Arg508 | 1.8 | Conventional covalent bond |
| 31 | Tyr371 | Arg508 | 1.8 | Conventional covalent bond |
| 32 | Ala375 | Lys505 | 2.1 | H-bond–van der Waals transition |
| 33 | Glu107 | Cys30 | 4.5 | Salt bridge |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, N.; Rabaan, A.A.; Alwashmi, A.S.S.; Albayat, H.; Mashraqi, M.M.; Alshehri, A.A.; Garout, M.; Abduljabbar, W.A.; Yusof, N.Y.; Yean, C.Y. Immunoinformatic Execution and Design of an Anti-Epstein–Barr Virus Vaccine with Multiple Epitopes Triggering Innate and Adaptive Immune Responses. Microorganisms 2023, 11, 2448. https://doi.org/10.3390/microorganisms11102448
Ahmed N, Rabaan AA, Alwashmi ASS, Albayat H, Mashraqi MM, Alshehri AA, Garout M, Abduljabbar WA, Yusof NY, Yean CY. Immunoinformatic Execution and Design of an Anti-Epstein–Barr Virus Vaccine with Multiple Epitopes Triggering Innate and Adaptive Immune Responses. Microorganisms. 2023; 11(10):2448. https://doi.org/10.3390/microorganisms11102448
Chicago/Turabian StyleAhmed, Naveed, Ali A. Rabaan, Ameen S. S. Alwashmi, Hawra Albayat, Mutaib M. Mashraqi, Ahmad A. Alshehri, Mohammed Garout, Wesam A. Abduljabbar, Nik Yusnoraini Yusof, and Chan Yean Yean. 2023. "Immunoinformatic Execution and Design of an Anti-Epstein–Barr Virus Vaccine with Multiple Epitopes Triggering Innate and Adaptive Immune Responses" Microorganisms 11, no. 10: 2448. https://doi.org/10.3390/microorganisms11102448