
Nanopore Sequencing Assessment of Bacterial Pathogens and Associated Antibiotic Resistance Genes in Environmental Samples


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
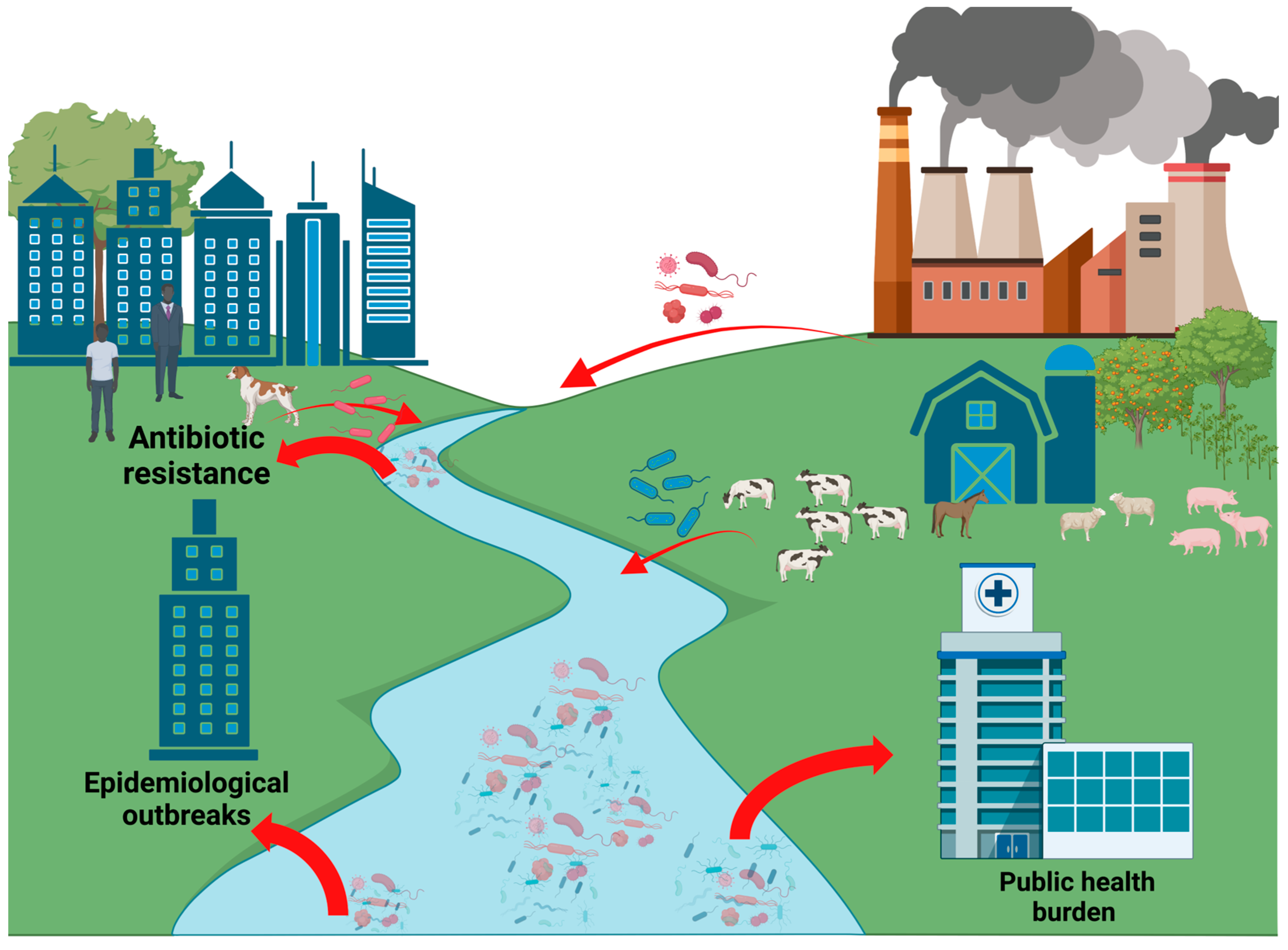
1.1. Relevance of Pathogenic Microorganisms in Municipal Waters
1.2. Sources of Antimicrobial Resistance Genes in Municipal Waters
- (a)
- The introduction of microorganisms that have already developed resistance to antibiotics through mutational mechanisms in response to antimicrobial factors. Genes resistant to aminoglycosides and β-lactams have been identified at all stages of farming in E. coli, a bacterium with a habitat in the intestinal tract [6].
- (b)
- The presence in the environment of the antimicrobial agents themselves. Only 30% of the antibiotics consumed are metabolized by users (humans/animals), whilst the highest amount is released in wastewater through feces and urine or by improper disposal, thus reaching bacterial communities with high density [7].
1.3. Detection Methods for AMR Surveillance and Research
2. Materials and Methods
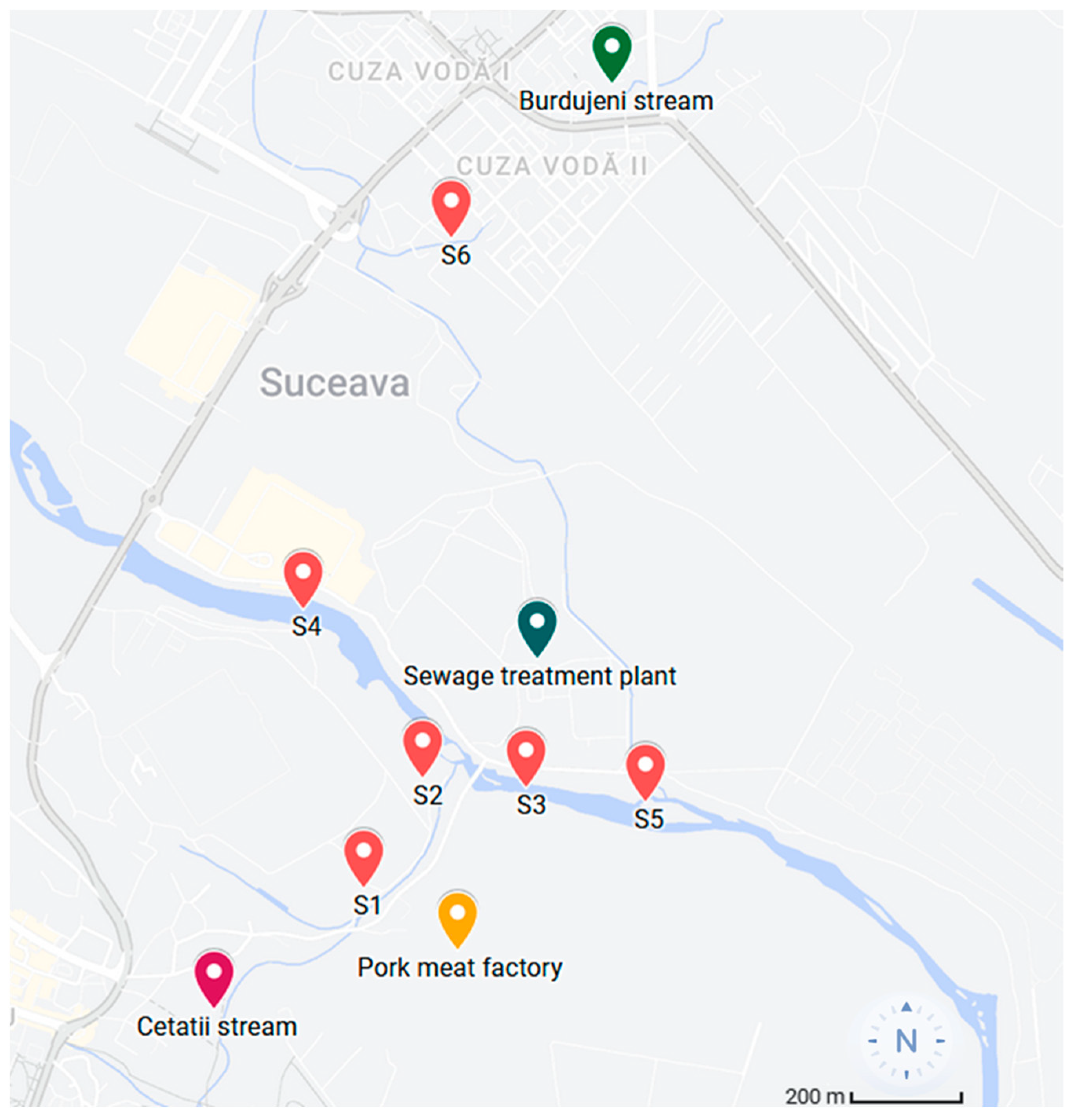
2.1. Sample Collection and Processing
2.2. DNA Extraction and Quantification
2.3. Library Preparation, Sequencing and Bioinformatic Analysis
2.4. Analysis of Diversity
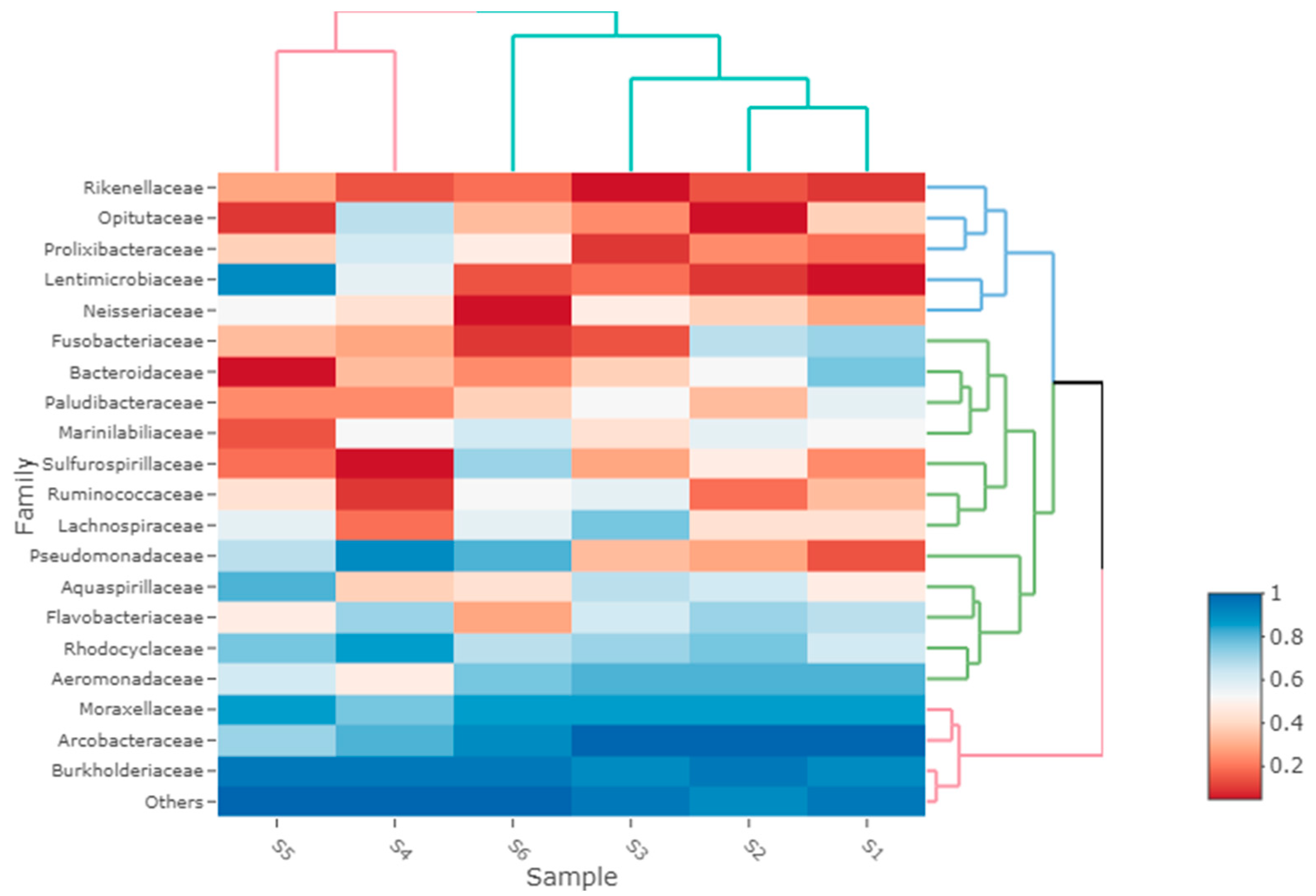
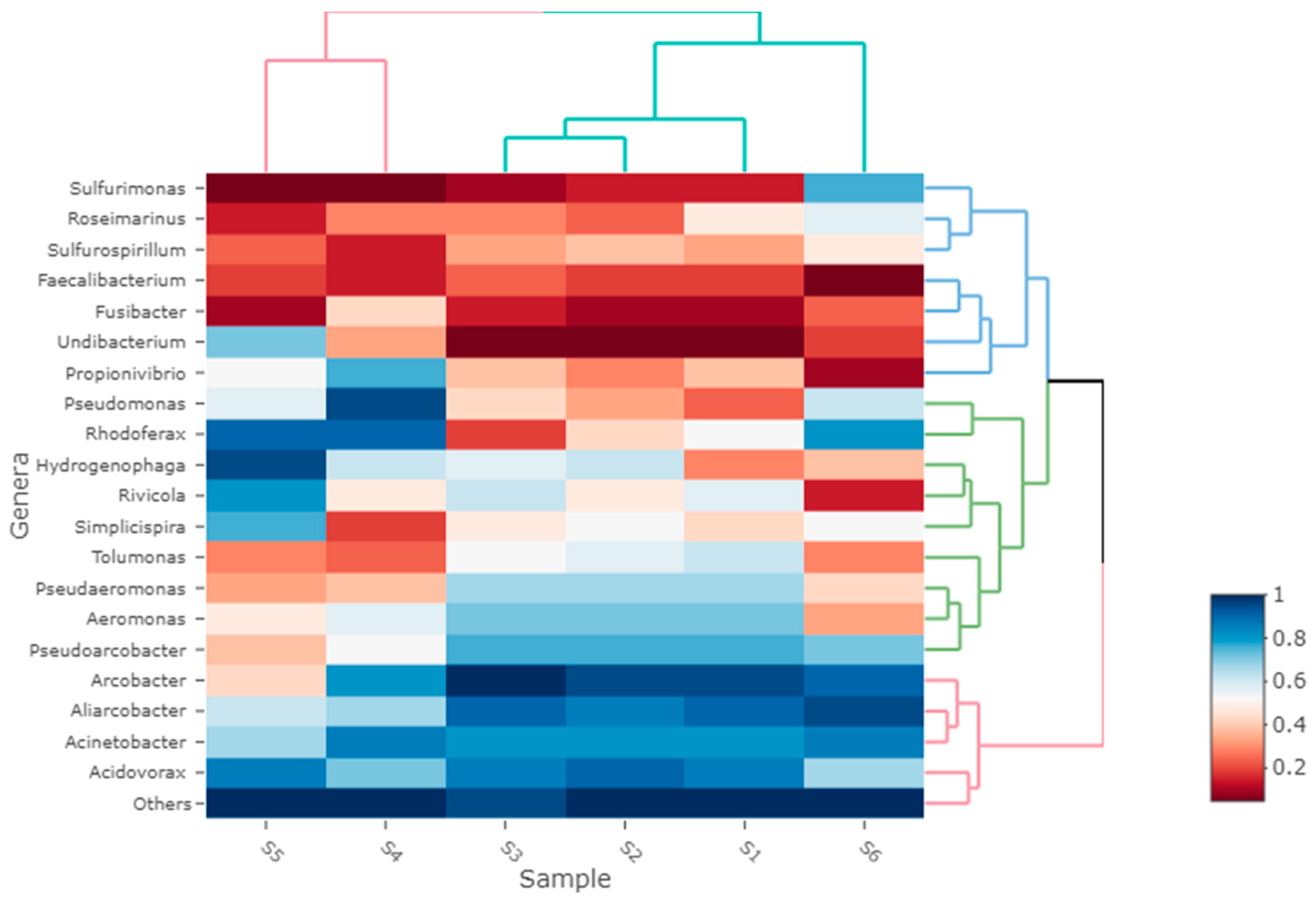
3. Results
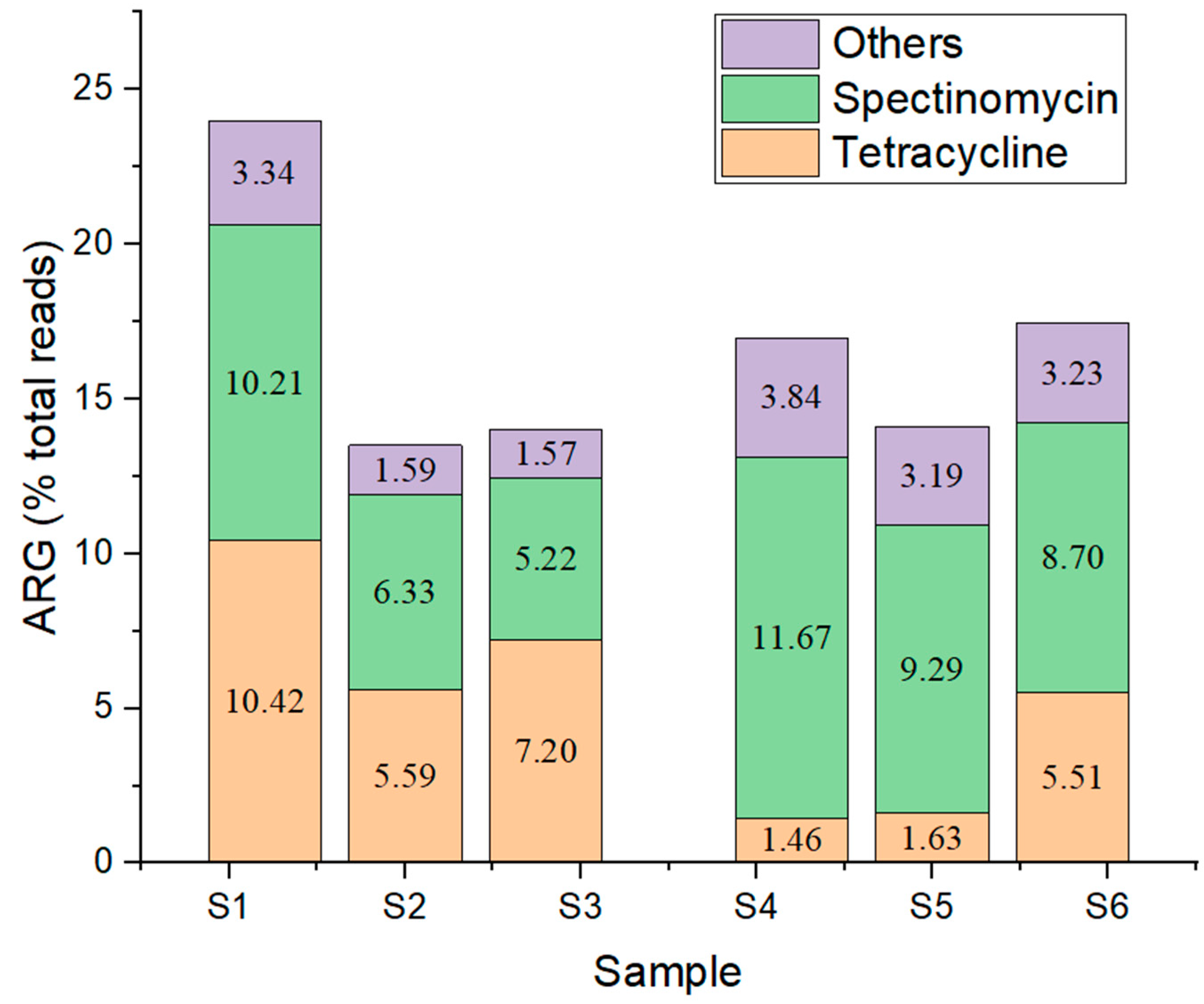
Antibiotic Resistance
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Varela, A.R.; Manaia, C.M. Human health implications of clinically relevant bacteria in wastewater habitats. Environ. Sci. Pollut. Res. 2013, 20, 3550–3569. [Google Scholar] [CrossRef]
- Chahal, C.; van den Akker, B.; Young, F.; Franco, C.; Blackbeard, J.; Monis, P. Pathogen and Particle Associations in Wastewater. Adv. Appl. Microbiol. 2016, 97, 63–119. [Google Scholar] [CrossRef]
- Cai, L.; Ju, F.; Zhang, T. Tracking human sewage microbiome in a municipal wastewater treatment plant. Appl. Microbiol. Biotechnol. 2014, 98, 3317–3326. [Google Scholar] [CrossRef]
- Ramírez-Castillo, F.Y.; Loera-Muro, A.; Jacques, M.; Garneau, P.; Avelar-González, F.J.; Harel, J.; Guerrero-Barrera, A.L. Waterborne Pathogens: Detection Methods and Challenges. Pathogens 2015, 4, 307–334. [Google Scholar] [CrossRef]
- Ajonina, C.; Buzie, C.; Rubiandini, R.H.; Otterpohl, R. Microbial Pathogens in Wastewater Treatment Plants (WWTP) in Hamburg. J. Toxicol. Environ. Health A 2015, 78, 381–387. [Google Scholar] [CrossRef]
- Lugsomya, K.; Yindee, J.; Niyomtham, W.; Tribuddharat, C.; Tummaruk, P.; Hampson, D.J.; Prapasarakul, N. Antimicrobial Resistance in Commensal Escherichia coli Isolated from Pigs and Pork Derived from Farms Either Routinely Using or Not Using In-Feed Antimicrobials. Microb. Drug Resist. 2018, 24, 1054–1066. [Google Scholar] [CrossRef]
- Nnadozie, C.F.; Kumari, S.; Bux, F. Status of pathogens, antibiotic resistance genes and antibiotic residues in wastewater treatment systems. Rev. Environ. Sci. Biotechnol. 2017, 16, 491–515. [Google Scholar] [CrossRef]
- Berendonk, T.U.; Manaia, C.M.; Merlin, C.; Fatta-Kassinos, D.; Cytryn, E.; Walsh, F.; Buergmann, H.; Sørum, H.; Norström, M.; Pons, M.-N.; et al. Tackling antibiotic resistance: The environmental framework. Nat. Rev. Microbiol. 2015, 13, 310–317. [Google Scholar] [CrossRef]
- Nguyen, A.Q.; Vu, H.P.; Nguyen, L.N.; Wang, Q.; Djordjevic, S.P.; Donner, E.; Yin, H.; Nghiem, L.D. Monitoring antibiotic resistance genes in wastewater treatment: Current strategies and future challenges. Sci. Total Environ. 2021, 783, 146964. [Google Scholar] [CrossRef]
- Wang, X.; Lan, B.; Fei, H.; Wang, S.; Zhu, G. Heavy metal could drive co-selection of antibiotic resistance in terrestrial subsurface soils. J. Hazard. Mater. 2021, 411, 124848. [Google Scholar] [CrossRef]
- Guidelines for Water Reuse; United States Environmental Protection Agency Washington. 2012. Available online: https://nepis.epa.gov/Exe/ZyPURL.cgi?Dockey=P100FS7K.txt (accessed on 7 April 2023).
- Bartram, J.; Ballance, R. (Eds.) Water Quality Monitoring: A Practical Guide to the Design and Implementation of Freshwater Quality Studies and Monitoring Programmes; CRC Press: Boca Raton, FL, USA, 1996. [Google Scholar]
- Garrido-Cardenas, J.A.; Polo-López, M.I.; Oller-Alberola, I. Advanced microbial analysis for wastewater quality monitoring: Metagenomics trend. Appl. Microbiol. Biotechnol. 2017, 101, 7445–7458. [Google Scholar] [CrossRef]
- ISO 13843:2017; Water Quality—Requirements for Establishing Performance Characteristics of Quantitative Microbiological Methods. ISO: Geneva, Switzerland, 2017.
- Ejeian, F.; Etedali, P.; Mansouri-Tehrani, H.-A.; Soozanipour, A.; Low, Z.-X.; Asadnia, M.; Taheri-Kafrani, A.; Razmjou, A. Biosensors for wastewater monitoring: A review. Biosens. Bioelectron. 2018, 118, 66–79. [Google Scholar] [CrossRef]
- Mao, K.; Zhang, H.; Pan, Y.; Yang, Z. Biosensors for wastewater-based epidemiology for monitoring public health. Water Res. 2021, 191, 116787. [Google Scholar] [CrossRef]
- Nkongolo, K.K.; Narendrula-Kotha, R. Advances in monitoring soil microbial community dynamic and function. J. Appl. Genet. 2020, 61, 249–263. [Google Scholar] [CrossRef]
- Lührig, K.; Canbäck, B.; Paul, C.J.; Johansson, T.; Persson, K.M.; Rådström, P. Bacterial Community Analysis of Drinking Water Biofilms in Southern Sweden. Microbes Environ. 2015, 30, 99–107. [Google Scholar] [CrossRef]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 319–338. [Google Scholar] [CrossRef]
- Wani, G.A.; Khan, M.A.; Dar, M.A.; Shah, M.A.; Reshi, Z.A. Next Generation High Throughput Sequencing to Assess Microbial Communities: An Application Based on Water Quality. Bull. Environ. Contam. Toxicol. 2021, 106, 727–733. [Google Scholar] [CrossRef]
- Sanz, J.L.; Köchling, T. Next-generation sequencing and waste/wastewater treatment: A comprehensive overview. Rev. Environ. Sci. Biotechnol. 2019, 18, 635–680. [Google Scholar] [CrossRef]
- Garner, E.; Davis, B.C.; Milligan, E.; Blair, M.F.; Keenum, I.; Maile-Moskowitz, A.; Pan, J.; Gnegy, M.; Liguori, K.; Gupta, S.; et al. Next generation sequencing approaches to evaluate water and wastewater quality. Water Res. 2021, 194, 116907. [Google Scholar] [CrossRef]
- Che, Y.; Xia, Y.; Liu, L.; Li, A.-D.; Yang, Y.; Zhang, T. Mobile antibiotic resistome in wastewater treatment plants revealed by Nanopore metagenomic sequencing. Microbiome 2019, 7, 44. [Google Scholar] [CrossRef]
- Yang, Y.; Che, Y.; Liu, L.; Wang, C.; Yin, X.; Deng, Y.; Yang, C.; Zhang, T. Rapid absolute quantification of pathogens and ARGs by nanopore sequencing. Sci. Total Environ. 2022, 809, 152190. [Google Scholar] [CrossRef]
- Acharya, K.; Khanal, S.; Pantha, K.; Amatya, N.; Davenport, R.J.; Werner, D. A comparative assessment of conventional and molecular methods, including MinION nanopore sequencing, for surveying water quality. Sci. Rep. 2019, 9, 15726. [Google Scholar] [CrossRef]
- Brandt, C.; Bongcam-Rudloff, E.; Müller, B. Abundance Tracking by Long-Read Nanopore Sequencing of Complex Microbial Communities in Samples from 20 Different Biogas/Wastewater Plants. Appl. Sci. 2020, 10, 7518. [Google Scholar] [CrossRef]
- Marin, C.; Marco-Jiménez, F.; Martínez-Priego, L.; De Marco-Romero, G.; Soriano-Chirona, V.; Lorenzo-Rebenaque, L.; D’auria, G. Rapid Oxford Nanopore Technologies MinION Sequencing Workflow for Campylobacter jejuni Identification in Broilers on Site—A Proof-of-Concept Study. Animals 2022, 12, 2065. [Google Scholar] [CrossRef]
- Ayiti, O.E.; Ayangbenro, A.S.; Babalola, O.O. 16S Amplicon Sequencing of Nitrifying Bacteria and Archaea Inhabiting Maize Rhizosphere and the Influencing Environmental Factors. Agriculture 2022, 12, 1328. [Google Scholar] [CrossRef]
- EPI2ME. Available online: https://epi2me.nanoporetech.com/ (accessed on 1 March 2023).
- Blankenberg, D.; Von Kuster, G.; Bouvier, E.; Baker, D.; Afgan, E.; Stoler, N.; Taylor, J.; Nekrutenko, A. Dissemination of scientific software with Galaxy ToolShed. Genome Biol. 2014, 15, 403. [Google Scholar] [CrossRef]
- Pascariu, G.; Dorobantu, M.; Stancu, E.; Perju, T.; Pintrijel, D.; Cazacu, A.; Minculescu, L.; Cioanger, I.; Cocheci, R. Etapa I/Infrastructuri Tehnice Majore. PATJ Suceava. 2019. Available online: http://www.cjsuceava.ro/ro/plan-de-amenajare-a-teritoriului-judetean-suceava (accessed on 10 April 2023).
- Venâncio, I.; Luís, Â.; Domingues, F.; Oleastro, M.; Pereira, L.; Ferreira, S. The Prevalence of Arcobacteraceae in Aquatic Environments: A Systematic Review and Meta-Analysis. Pathogens 2022, 11, 244. [Google Scholar] [CrossRef]
- Levican, A.; Collado, L.; Figueras, M.J. Arcobacter cloacae sp. nov. and Arcobacter suis sp. nov., two new species isolated from food and sewage. Syst. Appl. Microbiol. 2013, 36, 22–27. [Google Scholar] [CrossRef]
- Konishi, N.; Okubo, T.; Yamaya, T.; Hayakawa, T.; Minamisawa, K. Nitrate Supply-Dependent Shifts in Communities of Root-Associated Bacteria in Arabidopsis. Microbes Environ. 2017, 32, 314–323. [Google Scholar] [CrossRef]
- Weinroth, M.D.; Thomas, K.M.; Doster, E.; Vikram, A.; Schmidt, J.W.; Arthur, T.M.; Wheeler, T.L.; Parker, J.K.; Hanes, A.S.; Alekoza, N.; et al. Resistomes and microbiome of meat trimmings and colon content from culled cows raised in conventional and organic production systems. Anim. Microbiome 2022, 4, 21. [Google Scholar] [CrossRef]
- Ferreira, S.; Queiroz, J.A.; Oleastro, M.; Domingues, F.C. Insights in the pathogenesis and resistance of Arcobacter: A review. Crit. Rev. Microbiol. 2015, 42, 364–383. [Google Scholar] [CrossRef]
- Müller, E.; Hotzel, H.; Ahlers, C.; Hänel, I.; Tomaso, H.; Abdel-Glil, M.Y. Genomic Analysis and Antimicrobial Resistance of Aliarcobacter cryaerophilus Strains from German Water Poultry. Front. Microbiol. 2020, 11, 1549. [Google Scholar] [CrossRef]
- Çelik, C.; Pınar, O.; Sipahi, N. The Prevalence of Aliarcobacter Species in the Fecal Microbiota of Farm Animals and Potential Effective Agents for Their Treatment: A Review of the Past Decade. Microorganisms 2022, 10, 2430. [Google Scholar] [CrossRef]
- Willems, A.; Gillis, M. Acidovorax. In Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2015; pp. 1–16. [Google Scholar] [CrossRef]
- Wong, D.; Nielsen, T.B.; Bonomo, R.A.; Pantapalangkoor, P.; Luna, B.; Spellberg, B. Clinical and Pathophysiological Overview of Acinetobacter Infections: A Century of Challenges. Clin. Microbiol. Rev. 2017, 30, 409–447. [Google Scholar] [CrossRef]
- Hiraishi, A.; Imhoff, J.F. Rhodoferax. In Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2015; pp. 1–11. [Google Scholar] [CrossRef]
- Jin, C.-Z.; Zhuo, Y.; Wu, X.; Ko, S.-R.; Li, T.; Jin, F.-J.; Ahn, C.-Y.; Oh, H.-M.; Lee, H.-G.; Jin, L. Genomic and Metabolic Insights into Denitrification, Sulfur Oxidation, and Multidrug Efflux Pump Mechanisms in the Bacterium Rhodoferax sediminis sp. nov. Microorganisms 2020, 8, 262. [Google Scholar] [CrossRef]
- Suhana, S.; Nutan, Y.; Anoop, R.M. Interactive potential of Pseudomonas species with plants. J. Appl. Biol. Biotechnol. 2020, 8, 101–111. [Google Scholar] [CrossRef]
- Hashmi, M.Z. (Ed.) Antibiotics and Antimicrobial Resistance Genes; Springer International Publishing: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Asfaw, T. Review on hospital wastewater as a source of emerging drug resistance pathogens. J. Res. Environ. Sci. Toxicol. 2018, 7, 47–52. [Google Scholar] [CrossRef]
- Goodlet, K.J.; Benhalima, F.Z.; Nailor, M.D. A Systematic Review of Single-Dose Aminoglycoside Therapy for Urinary Tract Infection: Is It Time To Resurrect an Old Strategy? Antimicrob. Agents Chemother. 2019, 63, 10–1128. [Google Scholar] [CrossRef]
- Varela, M.F.; Stephen, J.; Lekshmi, M.; Ojha, M.; Wenzel, N.; Sanford, L.M.; Hernandez, A.J.; Parvathi, A.; Kumar, S.H. Bacterial Resistance to Antimicrobial Agents. Antibiotics 2021, 10, 593. [Google Scholar] [CrossRef]
- Guo, X.; Tang, N.; Lei, H.; Fang, Q.; Liu, L.; Zhou, Q.; Song, C. Metagenomic Analysis of Antibiotic Resistance Genes in Untreated Wastewater From Three Different Hospitals. Front. Microbiol. 2021, 12, 709051. [Google Scholar] [CrossRef]
- García-Solache, M.; Rice, L.B. The Enterococcus: A Model of Adaptability to Its Environment. Clin. Microbiol. Rev. 2019, 32, e00058-18. [Google Scholar] [CrossRef]
- Nessar, R.; Reyrat, J.M.; Murray, A.; Gicquel, B. Genetic analysis of new 16S rRNA mutations conferring aminoglycoside resistance in Mycobacterium abscessus. J. Antimicrob. Chemother. 2011, 66, 1719–1724. [Google Scholar] [CrossRef]
- Barton, M. Impact of antibiotic use in the swine industry. Curr. Opin. Microbiol. 2014, 19, 9–15. [Google Scholar] [CrossRef]
- Cromwell, G.L. Why and how antibiotics are used in swine production. Anim. Biotechnol. 2002, 13, 7–27. [Google Scholar] [CrossRef]
- Samreen; Ahmad, I.; Malak, H.A.; Abulreesh, H.H. Environmental antimicrobial resistance and its drivers: A potential threat to public health. J. Glob. Antimicrob. Resist. 2021, 27, 101–111. [Google Scholar] [CrossRef]
- Fang, L.; Chen, C.; Cui, C.; Li, X.; Zhang, Y.; Liao, X.; Sun, J.; Liu, Y. Emerging High-Level Tigecycline Resistance: Novel Tetracycline Destructases Spread via the Mobile Tet(X). BioEssays 2020, 42, e2000014. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobiuc, A.; Pavăl, N.-E.; Dimian, M.; Covașă, M. Nanopore Sequencing Assessment of Bacterial Pathogens and Associated Antibiotic Resistance Genes in Environmental Samples. Microorganisms 2023, 11, 2834. https://doi.org/10.3390/microorganisms11122834
Lobiuc A, Pavăl N-E, Dimian M, Covașă M. Nanopore Sequencing Assessment of Bacterial Pathogens and Associated Antibiotic Resistance Genes in Environmental Samples. Microorganisms. 2023; 11(12):2834. https://doi.org/10.3390/microorganisms11122834
Chicago/Turabian StyleLobiuc, Andrei, Naomi-Eunicia Pavăl, Mihai Dimian, and Mihai Covașă. 2023. "Nanopore Sequencing Assessment of Bacterial Pathogens and Associated Antibiotic Resistance Genes in Environmental Samples" Microorganisms 11, no. 12: 2834. https://doi.org/10.3390/microorganisms11122834
APA StyleLobiuc, A., Pavăl, N.-E., Dimian, M., & Covașă, M. (2023). Nanopore Sequencing Assessment of Bacterial Pathogens and Associated Antibiotic Resistance Genes in Environmental Samples. Microorganisms, 11(12), 2834. https://doi.org/10.3390/microorganisms11122834