
Targeting Viral Transcription for HIV Cure Strategies

 , and
, and 
Abstract
:1. Introduction
2. Cellular and Viral Factors Regulating HIV Transcription and Nuclear Export
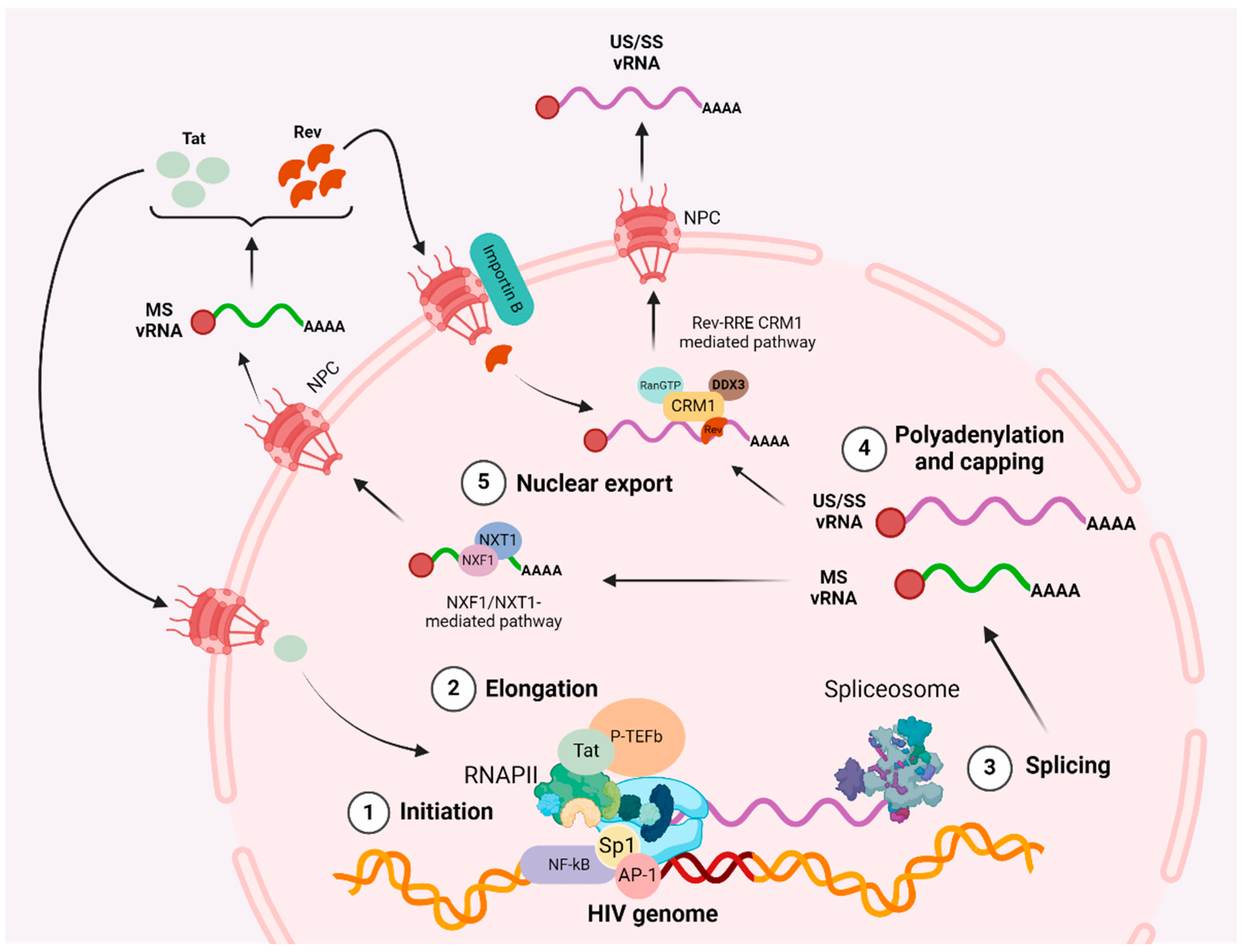
2.1. Initiation
2.2. Elongation
2.3. Polyadenylation
2.4. Multiple Splicing
2.5. Nuclear Export
2.6. Repressive Transcriptional Factors
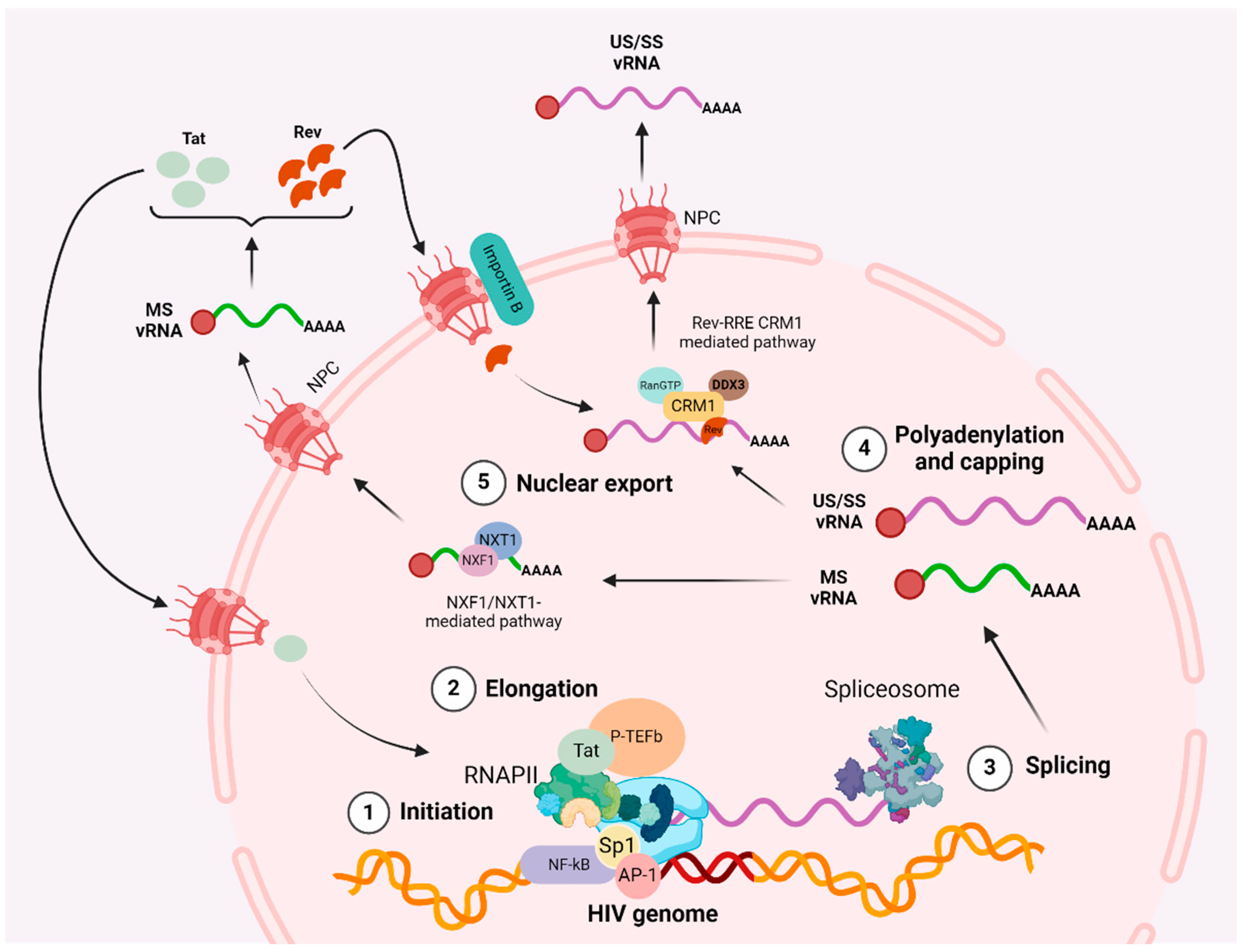
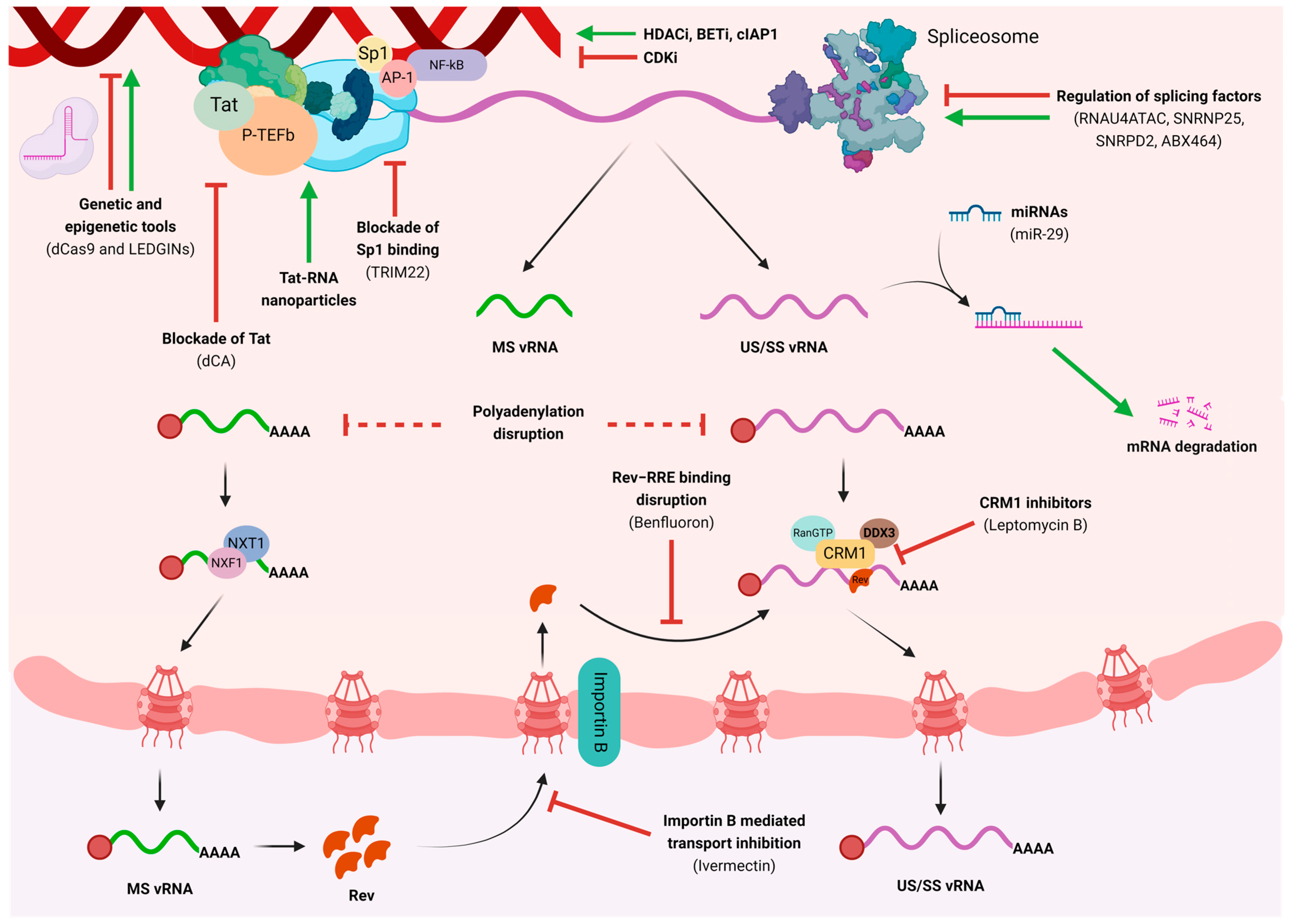
3. Pharmacological Disruption of HIV Transcription and Nuclear Export
4. Genetic and Epigenetic Modulation to Impact HIV Transcription
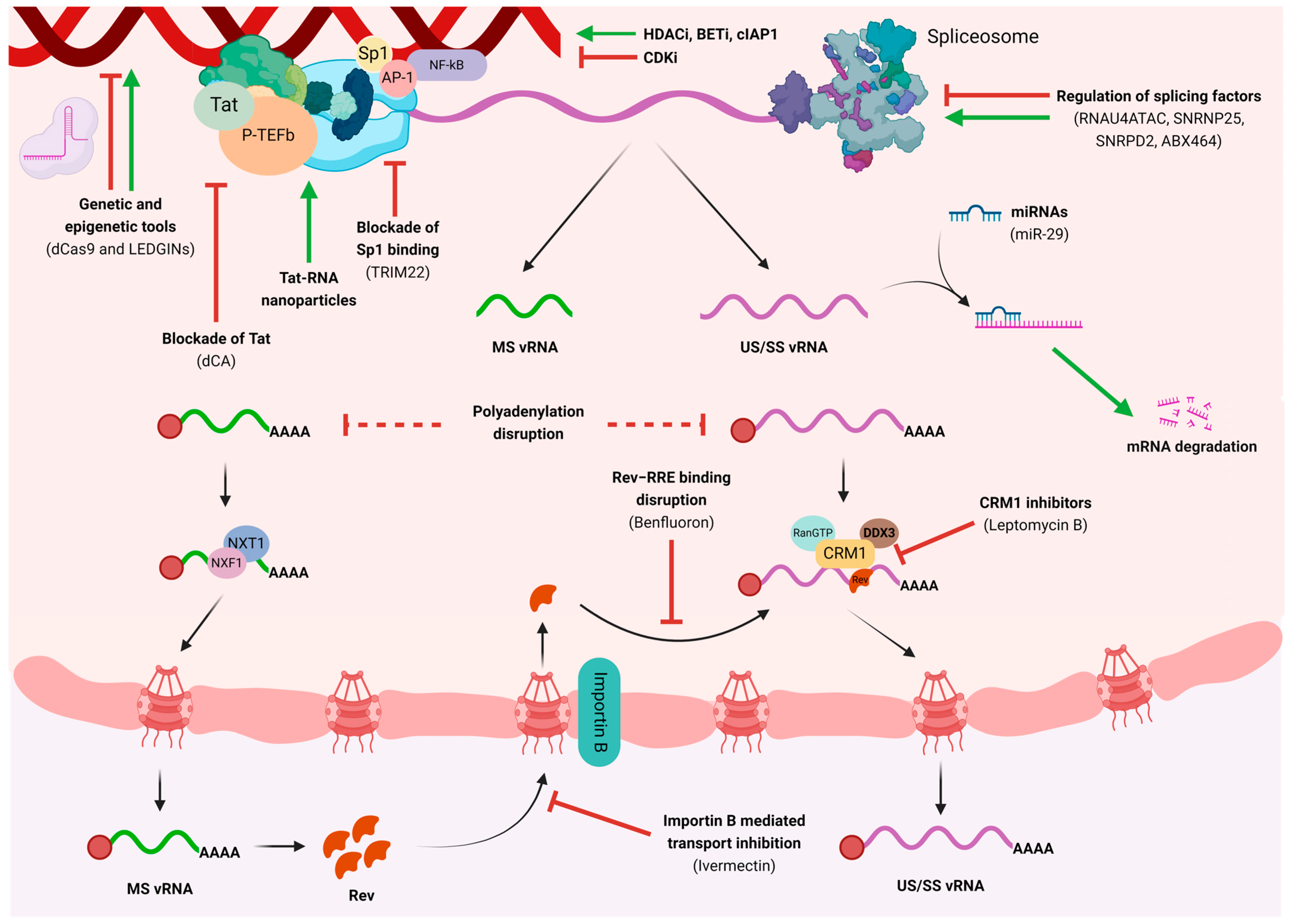
5. What Can We Learn from HIV-2?
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). HIV Statistics, Globally and by WHO Region, 2023; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Joint United Nations Programme on HIV/AIDS. The Path that Ends AIDS: UNAIDS Global AIDS Update 2023; United Nations: New York, NY, USA, 2023. [Google Scholar]
- Crespo, R.; Rao, S.; Mahmoudi, T. HibeRNAtion: HIV-1 RNA Metabolism and Viral Latency. Front. Cell. Infect. Microbiol. 2022, 12, 855092. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef]
- Lu, Y.; Bohn-Wippert, K.; Pazerunas, P.J.; Moy, J.M.; Singh, H.; Dar, R.D. Screening for gene expression fluctuations reveals latency-promoting agents of HIV. Proc. Natl. Acad. Sci. USA 2021, 118, e2012191118. [Google Scholar] [CrossRef]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a “Block-and-Lock” Strategy for HIV-1 Treatment. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, M.; Karn, J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef]
- Vargas, B.; Giacobbi, N.S.; Sanyal, A.; Venkatachari, N.J.; Han, F.; Gupta, P.; Sluis-Cremer, N. Inhibitors of Signaling Pathways That Block Reversal of HIV-1 Latency. Antimicrob. Agents Chemother. 2019, 63, e01744-18. [Google Scholar] [CrossRef] [PubMed]
- Mediouni, S.; Chinthalapudi, K.; Ekka, M.K.; Usui, I.; Jablonski, J.A.; Clementz, M.A.; Mousseau, G.; Nowak, J.; Macherla, V.R.; Beverage, J.N.; et al. Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat. MBio 2019, 10, e02662-18. [Google Scholar] [CrossRef]
- Dar, R.D.; Hosmane, N.N.; Arkin, M.R.; Siliciano, R.F.; Weinberger, L.S. Screening for noise in gene expression identifies drug synergies. Science 2014, 344, 1392–1396. [Google Scholar] [CrossRef]
- Balachandran, A.; Wong, R.; Stoilov, P.; Pan, S.; Blencowe, B.; Cheung, P.; Harrigan, P.R.; Cochrane, A. Identification of small molecule modulators of HIV-1 Tat and Rev protein accumulation. Retrovirology 2017, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Delannoy, A.; Poirier, M.; Bell, B. Cat and Mouse: HIV Transcription in Latency, Immune Evasion and Cure/Remission Strategies. Viruses 2019, 11, 269. [Google Scholar] [CrossRef]
- Jean, M.J.; Fiches, G.; Hayashi, T.; Zhu, J. Current Strategies for Elimination of HIV-1 Latent Reservoirs Using Chemical Compounds Targeting Host and Viral Factors. AIDS Res. Hum. Retroviruses 2019, 35, 1–24. [Google Scholar] [CrossRef] [PubMed]
- el Kharroubi, A.; Verdin, E. Protein-DNA interactions within DNase I-hypersensitive sites located downstream of the HIV-1 promoter. J. Biol. Chem. 1994, 269, 19916–19924. [Google Scholar] [CrossRef]
- Van Lint, C.; Amella, C.A.; Emiliani, S.; John, M.; Jie, T.; Verdin, E. Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J. Virol. 1997, 71, 6113–6127. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Latchman, D.S. The octamer-binding proteins Oct-1 and Oct-2 repress the HIV long terminal repeat promoter and its transactivation by Tat. Biochem. J. 1997, 322 Pt 1, 155–158. [Google Scholar] [CrossRef]
- Ruocco, M.R.; Chen, X.; Ambrosino, C.; Dragonetti, E.; Liu, W.; Mallardo, M.; De Falco, G.; Palmieri, C.; Franzoso, G.; Quinto, I.; et al. Regulation of HIV-1 Long Terminal Repeats by Interaction of C/EBP(NF-IL6) and NF-κB/Rel Transcription Factors. J. Biol. Chem. 1996, 271, 22479–22486. [Google Scholar] [CrossRef] [PubMed]
- Montano, M.A.; Kripke, K.; Norina, C.D.; Achacoso, P.; Herzenberg, L.A.; Roy, A.L.; Nolan, G.P. NF-kappa B homodimer binding within the HIV-1 initiator region and interactions with TFII-I. Proc. Natl. Acad. Sci. USA 1996, 93, 12376–12381. [Google Scholar] [CrossRef]
- Dutilleul, A.; Rodari, A.; Van Lint, C. Depicting HIV-1 Transcriptional Mechanisms: A Summary of What We Know. Viruses 2020, 12, 1385. [Google Scholar] [CrossRef]
- Parada, C.A.; Roeder, R.G. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature 1996, 384, 375–378. [Google Scholar] [CrossRef]
- Molle, D.; Maiuri, P.; Boireau, S.; Bertrand, E.; Knezevich, A.; Marcello, A.; Basyuk, E. A real-time view of the TAR:Tat:P-TEFb complex at HIV-1 transcription sites. Retrovirology 2007, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J. Putting an “End” to HIV mRNAs: Capping and polyadenylation as potential therapeutic targets. AIDS Res. Ther. 2013, 10, 31. [Google Scholar] [CrossRef]
- Shi, Y.; Di Giammartino, D.C.; Taylor, D.; Sarkeshik, A.; Rice, W.J.; Yates, J.R., 3rd; Frank, J.; Manley, J.L. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol. Cell 2009, 33, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B. HIV-1 as RNA evolution machine. RNA Biol. 2011, 8, 225–229. [Google Scholar] [CrossRef]
- Telwatte, S.; Morón-López, S.; Aran, D.; Kim, P.; Hsieh, C.; Joshi, S.; Montano, M.; Greene, W.C.; Butte, A.J.; Wong, J.K.; et al. Heterogeneity in HIV and cellular transcription profiles in cell line models of latent and productive infection: Implications for HIV latency. Retrovirology 2019, 16, 32. [Google Scholar] [CrossRef]
- Stoltzfus, C.M. Chapter 1. Regulation of HIV-1 alternative RNA splicing and its role in virus replication. Adv. Virus Res. 2009, 74, 1–40. [Google Scholar] [CrossRef]
- Crespo, R.; Ne, E.; Reinders, J.; Meier, J.I.J.; Li, C.; Jansen, S.; Górska, A.; Koçer, S.; Kan, T.W.; Doff, W.; et al. PCID2 dysregulates transcription and viral RNA processing to promote HIV-1 latency. iScience 2024, 27, 109152. [Google Scholar] [CrossRef]
- Moron-Lopez, S.; Telwatte, S.; Sarabia, I.; Battivelli, E.; Montano, M.; Macedo, A.B.; Aran, D.; Butte, A.J.; Jones, R.B.; Bosque, A.; et al. Human splice factors contribute to latent HIV infection in primary cell models and blood CD4+ T cells from ART-treated individuals. PLoS Pathog. 2020, 16, e1009060. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.; Durbin, R. A computational scan for U12-dependent introns in the human genome sequence. Nucleic Acids Res. 2001, 29, 4006–4013. [Google Scholar] [CrossRef]
- Will, C.L.; Lührmann, R. Splicing of a rare class of introns by the U12-dependent spliceosome. Biol. Chem. 2005, 386, 713–724. [Google Scholar] [CrossRef]
- Patel, A.A.; Steitz, J.A. Splicing double: Insights from the second spliceosome. Nat. Rev. Mol. Cell Biol. 2003, 4, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Nuclear mRNA export: Insights from virology. Trends Biochem. Sci. 2003, 28, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Köhler, A.; Hurt, E. Exporting RNA from the nucleus to the cytoplasm. Nat. Rev. Mol. Cell Biol. 2007, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.R.; Percipalle, P. Interactions between HIV Rev and nuclear import and export factors: The Rev nuclear localisation signal mediates specific binding to human importin-beta. J. Mol. Biol. 1997, 274, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Campbell, M.; Nasioulas, G.; Harrison, J.; Felber, B.K.; Pavlakis, G.N. Mutational inactivation of an inhibitory sequence in human immunodeficiency virus type 1 results in Rev-independent gag expression. J. Virol. 1992, 66, 7176–7182. [Google Scholar] [CrossRef]
- Schwartz, S.; Felber, B.K.; Pavlakis, G.N. Distinct RNA sequences in the gag region of human immunodeficiency virus type 1 decrease RNA stability and inhibit expression in the absence of Rev protein. J. Virol. 1992, 66, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Schiralli Lester, G.M.; Henderson, A.J. Mechanisms of HIV Transcriptional Regulation and Their Contribution to Latency. Mol. Biol. Int. 2012, 2012, 614120. [Google Scholar] [CrossRef]
- Tissot, C.; Mechti, N. Molecular cloning of a new interferon-induced factor that represses human immunodeficiency virus type 1 long terminal repeat expression. J. Biol. Chem. 1995, 270, 14891–14898. [Google Scholar] [CrossRef]
- Pagani, I.; Poli, G.; Vicenzi, E. TRIM22. A Multitasking Antiviral Factor. Cells 2021, 10, 1864. [Google Scholar] [CrossRef]
- Kajaste-Rudnitski, A.; Marelli, S.S.; Pultrone, C.; Pertel, T.; Uchil, P.D.; Mechti, N.; Mothes, W.; Poli, G.; Luban, J.; Vicenzi, E. TRIM22 inhibits HIV-1 transcription independently of its E3 ubiquitin ligase activity, Tat, and NF-kappaB-responsive long terminal repeat elements. J. Virol. 2011, 85, 5183–5196. [Google Scholar] [CrossRef]
- Singh, R.; Gaiha, G.; Werner, L.; McKim, K.; Mlisana, K.; Luban, J.; Walker, B.D.; Karim, S.S.A.; Brass, A.L.; Ndung’u, T. Association of TRIM22 with the type 1 interferon response and viral control during primary HIV-1 infection. J. Virol. 2011, 85, 208–216. [Google Scholar] [CrossRef]
- Morón-López, S.; Gómez-Mora, E.; Salgado, M.; Ouchi, D.; Puertas, M.C.; Urrea, V.; Navarro, J.; Jou, A.; Pérez, M.; Tural, C.; et al. Short-term Treatment With Interferon Alfa Diminishes Expression of HIV-1 and Reduces CD4+ T-Cell Activation in Patients Coinfected With HIV and Hepatitis C Virus and Receiving Antiretroviral Therapy. J. Infect. Dis. 2016, 213, 1008–1012. [Google Scholar] [CrossRef]
- Sanchez, J.G.; Okreglicka, K.; Chandrasekaran, V.; Welker, J.M.; Sundquist, W.I.; Pornillos, O. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc. Natl. Acad. Sci. USA 2014, 111, 2494–2499. [Google Scholar] [CrossRef] [PubMed]
- Marban, C.; Suzanne, S.; Dequiedt, F.; de Walque, S.; Redel, L.; Van Lint, C.; Aunis, D.; Rohr, O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Cismasiu, V.B.; Paskaleva, E.; Suman Daya, S.; Canki, M.; Duus, K.; Avram, D. BCL11B is a general transcriptional repressor of the HIV-1 long terminal repeat in T lymphocytes through recruitment of the NuRD complex. Virology 2008, 380, 173–181. [Google Scholar] [CrossRef]
- Pedro, K.D.; Agosto, L.M.; Sewell, J.A.; Eberenz, K.A.; He, X.; Fuxman Bass, J.I.; Henderson, A.J. A functional screen identifies transcriptional networks that regulate HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2012835118. [Google Scholar] [CrossRef]
- Natarajan, M.; Schiralli Lester, G.M.; Lee, C.; Missra, A.; Wasserman, G.A.; Steffen, M.; Gilmour, D.S.; Henderson, A.J. Negative elongation factor (NELF) coordinates RNA polymerase II pausing, premature termination, and chromatin remodeling to regulate HIV transcription. J. Biol. Chem. 2013, 288, 25995–26003. [Google Scholar] [CrossRef] [PubMed]
- Bruce, J.W.; Bracken, M.; Evans, E.; Sherer, N.; Ahlquist, P. ZBTB2 represses HIV-1 transcription and is regulated by HIV-1 Vpr and cellular DNA damage responses. PLoS Pathog. 2021, 17, e1009364. [Google Scholar] [CrossRef]
- Buratowski, S. Connections between mRNA 3′ end processing and transcription termination. Curr. Opin. Cell Biol. 2005, 17, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Rosonina, E.; Kaneko, S.; Manley, J.L. Terminating the transcript: Breaking up is hard to do. Genes Dev. 2006, 20, 1050–1056. [Google Scholar] [CrossRef]
- Zhang, Z.; Klatt, A.; Henderson, A.J.; Gilmour, D.S. Transcription termination factor Pcf11 limits the processivity of Pol II on an HIV provirus to repress gene expression. Genes Dev. 2007, 21, 1609–1614. [Google Scholar] [CrossRef]
- Ait Said, M.; Bejjani, F.; Abdouni, A.; Ségéral, E.; Emiliani, S. Premature transcription termination complex proteins PCF11 and WDR82 silence HIV-1 expression in latently infected cells. Proc. Natl. Acad. Sci. USA 2023, 120, e2313356120. [Google Scholar] [CrossRef]
- Matkovic, R.; Morel, M.; Lanciano, S.; Larrous, P.; Martin, B.; Bejjani, F.; Vauthier, V.; Hansen, M.M.K.; Emiliani, S.; Cristofari, G.; et al. TASOR epigenetic repressor cooperates with a CNOT1 RNA degradation pathway to repress HIV. Nat. Commun. 2022, 13, 66. [Google Scholar] [CrossRef]
- Archin, N.M.; Bateson, R.; Tripathy, M.K.; Crooks, A.M.; Yang, K.-H.; Dahl, N.P.; Kearney, M.F.; Anderson, E.M.; Coffin, J.M.; Strain, M.C.; et al. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. J. Infect. Dis. 2014, 210, 728–735. [Google Scholar] [CrossRef]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [PubMed]
- Leth, S.; Schleimann, M.H.; Nissen, S.K.; Højen, J.F.; Olesen, R.; Graversen, M.E.; Jørgensen, S.; Kjær, A.S.; Denton, P.W.; Mørk, A.; et al. Combined effect of Vacc-4x, recombinant human granulocyte macrophage colony-stimulating factor vaccination, and romidepsin on the HIV-1 reservoir (REDUC): A single-arm, phase 1B/2A trial. Lancet HIV 2016, 3, e463–e472. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef] [PubMed]
- Søgaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B.M. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J. Biol. Chem. 2012, 287, 36609–36616. [Google Scholar] [CrossRef]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.-C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef]
- Contreras, X.; Schweneker, M.; Chen, C.-S.; McCune, J.M.; Deeks, S.G.; Martin, J.; Peterlin, B.M. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 2009, 284, 6782–6789. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.A.; Tolstrup, M.; Winckelmann, A.; Østergaard, L.; Søgaard, O.S. Eliminating the latent HIV reservoir by reactivation strategies: Advancing to clinical trials. Hum. Vaccines Immunother. 2013, 9, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.G.; Chiang, V.; Fyne, E.; Balakrishnan, M.; Barnes, T.; Graupe, M.; Hesselgesser, J.; Irrinki, A.; Murry, J.P.; Stepan, G.; et al. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog. 2014, 10, e1004071. [Google Scholar] [CrossRef] [PubMed]
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature 2020, 578, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Pache, L.; Marsden, M.D.; Teriete, P.; Portillo, A.J.; Heimann, D.; Kim, J.T.; Soliman, M.S.A.; Dimapasoc, M.; Carmona, C.; Celeridad, M.; et al. Pharmacological Activation of Non-canonical NF-κB Signaling Activates Latent HIV-1 Reservoirs In Vivo. Cell Rep. Med. 2020, 1, 100037. [Google Scholar] [CrossRef] [PubMed]
- Blazkova, J.; Chun, T.-W.; Belay, B.W.; Murray, D.; Justement, J.S.; Funk, E.K.; Nelson, A.; Hallahan, C.W.; Moir, S.; Wender, P.A.; et al. Effect of histone deacetylase inhibitors on HIV production in latently infected, resting CD4(+) T cells from infected individuals receiving effective antiretroviral therapy. J. Infect. Dis. 2012, 206, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Moron-Lopez, S.; Kim, P.; Søgaard, O.S.; Tolstrup, M.; Wong, J.K.; Yukl, S.A. Characterization of the HIV-1 transcription profile after romidepsin administration in ART-suppressed individuals. AIDS 2019, 33, 425–431. [Google Scholar] [CrossRef]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Telwatte, S.; Joshi, S.K.; Vu, M.; Lampiris, H.; Wong, J.K. HIV latency in isolated patient CD4(+) T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med. 2018, 10, eaap9927. [Google Scholar] [CrossRef]
- Lassen, K.G.; Ramyar, K.X.; Bailey, J.R.; Zhou, Y.; Siliciano, R.F. Nuclear Retention of Multiply Spliced HIV-1 RNA in Resting CD4+ T Cells. PLoS Pathog. 2006, 2, e68. [Google Scholar] [CrossRef]
- Pardons, M.; Cole, B.; Lambrechts, L.; van Snippenberg, W.; Rutsaert, S.; Noppe, Y.; De Langhe, N.; Dhondt, A.; Vega, J.; Eyassu, F.; et al. Potent latency reversal by Tat RNA-containing nanoparticle enables multi-omic analysis of the HIV-1 reservoir. Nat. Commun. 2023, 14, 8397. [Google Scholar] [CrossRef] [PubMed]
- Van Gulck, E.; Pardons, M.; Nijs, E.; Verheyen, N.; Dockx, K.; Van Den Eynde, C.; Battivelli, E.; Vega, J.; Florence, E.; Autran, B.; et al. A truncated HIV Tat demonstrates potent and specific latency reversal activity. Antimicrob. Agents Chemother. 2023, 67, e0041723. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, I.M.; Wadia, J.S.; Dowdy, S.F. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release 2005, 102, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Molyer, B.; Kumar, A.; Angel, J.B. SMAC Mimetics as Therapeutic Agents in HIV Infection. Front. Immunol. 2021, 12, 780400. [Google Scholar] [CrossRef] [PubMed]
- Mori, L.; Valente, S.T. Key Players in HIV-1 Transcriptional Regulation: Targets for a Functional Cure. Viruses 2020, 12, 529. [Google Scholar] [CrossRef] [PubMed]
- Moranguinho, I.; Valente, S.T. Block-And-Lock: New Horizons for a Cure for HIV-1. Viruses 2020, 12, 1443. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Li, D.; Lin, M.-H.; Li, L.; Harrich, D. Tat-Based Therapies as an Adjuvant for an HIV-1 Functional Cure. Viruses 2020, 12, 415. [Google Scholar] [CrossRef]
- Mediouni, S.; Kessing, C.F.; Jablonski, J.A.; Thenin-Houssier, S.; Clementz, M.; Kovach, M.D.; Mousseau, G.; de Vera, I.M.S.; Li, C.; Kojetin, D.J.; et al. The Tat inhibitor didehydro-cortistatin A suppresses SIV replication and reactivation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 8280–8293. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. MBio 2015, 6, e00465. [Google Scholar] [CrossRef]
- Vigón, L.; Martínez-Román, P.; Rodríguez-Mora, S.; Torres, M.; Puertas, M.C.; Mateos, E.; Salgado, M.; Navarro, A.; Sánchez-Conde, M.; Ambrosioni, J.; et al. Provirus reactivation is impaired in HIV-1 infected individuals on treatment with dasatinib and antiretroviral therapy. Biochem. Pharmacol. 2021, 192, 114666. [Google Scholar] [CrossRef]
- Salgado, M.; Martinez-Picado, J.; Gálvez, C.; Rodríguez-Mora, S.; Rivaya, B.; Urrea, V.; Mateos, E.; Alcamí, J.; Coiras, M. Dasatinib protects humanized mice from acute HIV-1 infection. Biochem. Pharmacol. 2020, 174, 113625. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.M.; Brumme, Z.L.; Sadowski, I. Small molecule inhibitors of transcriptional cyclin-dependent kinases impose HIV-1 latency, presenting “block and lock” treatment strategies. Antimicrob. Agents Chemother. 2024, 68, e0107223. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.K.; Lykke-Andersen, S.; Jensen, T.H. Promoter-proximal polyadenylation sites reduce transcription activity. Genes Dev. 2012, 26, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Hauptman, P.J.; Kelly, R.A. Digitalis. Circulation 1999, 99, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Luk, B.T.; Hamidy, M.; Zhang, L.; Spector, S.A. Induction of a Na(+)/K(+)-ATPase-dependent form of autophagy triggers preferential cell death of human immunodeficiency virus type-1-infected macrophages. Autophagy 2018, 14, 1359–1375. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.-H.J.; Jenike, K.M.; Calvi, R.M.; Chiarella, J.; Hoh, R.; Deeks, S.G.; Ho, Y.-C. Filgotinib suppresses HIV-1-driven gene transcription by inhibiting HIV-1 splicing and T cell activation. J. Clin. Investig. 2020, 130, 4969–4984. [Google Scholar] [CrossRef]
- Wagstaff, K.M.; Sivakumaran, H.; Heaton, S.M.; Harrich, D.; Jans, D.A. Ivermectin is a specific inhibitor of importin α/β-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem. J. 2012, 443, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.W.; Balachandran, A.; Haaland, M.; Stoilov, P.; Cochrane, A. Characterization of novel inhibitors of HIV-1 replication that function via alteration of viral RNA processing and rev function. Nucleic Acids Res. 2013, 41, 9471–9483. [Google Scholar] [CrossRef]
- Wolff, B.; Sanglier, J.J.; Wang, Y. Leptomycin B is an inhibitor of nuclear export: Inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol. 1997, 4, 139–147. [Google Scholar] [CrossRef]
- Prado, S.; Beltrán, M.; Moreno, Á.; Bedoya, L.M.; Alcamí, J.; Gallego, J. A small-molecule inhibitor of HIV-1 Rev function detected by a diversity screen based on RRE-Rev interference. Biochem. Pharmacol. 2018, 156, 68–77. [Google Scholar] [CrossRef]
- Daelemans, D.; Afonina, E.; Nilsson, J.; Werner, G.; Kjems, J.; De Clercq, E.; Pavlakis, G.N.; Vandamme, A.-M. A synthetic HIV-1 Rev inhibitor interfering with the CRM1-mediated nuclear export. Proc. Natl. Acad. Sci. USA 2002, 99, 14440–14445. [Google Scholar] [CrossRef]
- Boons, E.; Vanstreels, E.; Jacquemyn, M.; Nogueira, T.C.; Neggers, J.E.; Vercruysse, T.; van den Oord, J.; Tamir, S.; Shacham, S.; Landesman, Y.; et al. Human Exportin-1 is a Target for Combined Therapy of HIV and AIDS Related Lymphoma. EBioMedicine 2015, 2, 1102–1113. [Google Scholar] [CrossRef]
- Chumillas, S.; Loharch, S.; Beltrán, M.; Szewczyk, M.P.; Bernal, S.; Puertas, M.C.; Martinez-Picado, J.; Alcamí, J.; Bedoya, L.M.; Marchán, V.; et al. Exploring the HIV-1 Rev Recognition Element (RRE)–Rev Inhibitory Capacity and Antiretroviral Action of Benfluron Analogs. Molecules 2023, 28, 7031. [Google Scholar] [CrossRef] [PubMed]
- Campos, N.; Myburgh, R.; Garcel, A.; Vautrin, A.; Lapasset, L.; Nadal, E.S.; Mahuteau-Betzer, F.; Najman, R.; Fornarelli, P.; Tantale, K.; et al. Long lasting control of viral rebound with a new drug ABX464 targeting Rev—Mediated viral RNA biogenesis. Retrovirology 2015, 12, 30. [Google Scholar] [CrossRef] [PubMed]
- Vautrin, A.; Manchon, L.; Garcel, A.; Campos, N.; Lapasset, L.; Laaref, A.M.; Bruno, R.; Gislard, M.; Dubois, E.; Scherrer, D.; et al. Both anti-inflammatory and antiviral properties of novel drug candidate ABX464 are mediated by modulation of RNA splicing. Sci. Rep. 2019, 9, 792. [Google Scholar] [CrossRef] [PubMed]
- Moron-Lopez, S.; Bernal, S.; Wong, J.K.; Martinez-Picado, J.; Yukl, S.A. ABX464 Decreases the Total Human Immunodeficiency Virus (HIV) Reservoir and HIV Transcription Initiation in CD4+ T Cells From Antiretroviral Therapy–Suppressed Individuals Living With HIV. Clin. Infect. Dis. 2022, 74, 2044–2049. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Shen, Y.; Yang, H.; Wang, Y.; Jiang, Z.; Yang, X.; Zhong, Y.; Pan, H.; Xu, J.; Lu, H.; et al. BET inhibitors RVX-208 and PFI-1 reactivate HIV-1 from latency. Sci. Rep. 2017, 7, 16646. [Google Scholar] [CrossRef]
- Zhang, X.-X.; Lin, J.; Liang, T.-Z.; Duan, H.; Tan, X.-H.; Xi, B.-M.; Li, L.; Liu, S.-W. The BET bromodomain inhibitor apabetalone induces apoptosis of latent HIV-1 reservoir cells following viral reactivation. Acta Pharmacol. Sin. 2019, 40, 98–110. [Google Scholar] [CrossRef]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef]
- Liang, T.; Zhang, X.; Lai, F.; Lin, J.; Zhou, C.; Xu, X.; Tan, X.; Liu, S.; Li, L. A novel bromodomain inhibitor, CPI-203, serves as an HIV-1 latency-reversing agent by activating positive transcription elongation factor b. Biochem. Pharmacol. 2019, 164, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, Z.; Reszka-Blanco, N.; Li, F.; Chi, L.; Ma, J.; Jeffrey, J.; Cheng, L.; Su, L. Specific Activation In Vivo of HIV-1 by a Bromodomain Inhibitor from Monocytic Cells in Humanized Mice under Antiretroviral Therapy. J. Virol. 2019, 93, e00233-19. [Google Scholar] [CrossRef]
- Abner, E.; Stoszko, M.; Zeng, L.; Chen, H.-C.; Izquierdo-Bouldstridge, A.; Konuma, T.; Zorita, E.; Fanunza, E.; Zhang, Q.; Mahmoudi, T.; et al. A New Quinoline BRD4 Inhibitor Targets a Distinct Latent HIV-1 Reservoir for Reactivation from Other “Shock” Drugs. J. Virol. 2018, 92, e02056-17. [Google Scholar] [CrossRef]
- Lu, P.; Qu, X.; Shen, Y.; Jiang, Z.; Wang, P.; Zeng, H.; Ji, H.; Deng, J.; Yang, X.; Li, X.; et al. The BET inhibitor OTX015 reactivates latent HIV-1 through P-TEFb. Sci. Rep. 2016, 6, 24100. [Google Scholar] [CrossRef]
- Zhao, M.; De Crignis, E.; Rokx, C.; Verbon, A.; van Gelder, T.; Mahmoudi, T.; Katsikis, P.D.; Mueller, Y.M. T cell toxicity of HIV latency reversing agents. Pharmacol. Res. 2019, 139, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liu, S.; Jean, M.; Simpson, S.; Huang, H.; Merkley, M.; Hayashi, T.; Kong, W.; Rodríguez-Sánchez, I.; Zhang, X.; et al. A Novel Bromodomain Inhibitor Reverses HIV-1 Latency through Specific Binding with BRD4 to Promote Tat and P-TEFb Association. Front. Microbiol. 2017, 8, 1035. [Google Scholar] [CrossRef]
- Rodríguez-Mora, S.; Spivak, A.M.; Szaniawski, M.A.; López-Huertas, M.R.; Alcamí, J.; Planelles, V.; Coiras, M. Tyrosine Kinase Inhibition: A New Perspective in the Fight against HIV. Curr. HIV/AIDS Rep. 2019, 16, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Reiser, K.; Mathys, L.; Curbo, S.; Pannecouque, C.; Noppen, S.; Liekens, S.; Engman, L.; Lundberg, M.; Balzarini, J.; Karlsson, A. The Cellular Thioredoxin-1/Thioredoxin Reductase-1 Driven Oxidoreduction Represents a Chemotherapeutic Target for HIV-1 Entry Inhibition. PLoS ONE 2016, 11, e0147773. [Google Scholar] [CrossRef]
- Didigu, C.A.; Wilen, C.B.; Wang, J.; Duong, J.; Secreto, A.J.; Danet-Desnoyers, G.A.; Riley, J.L.; Gregory, P.D.; June, C.H.; Holmes, M.C.; et al. Simultaneous zinc-finger nuclease editing of the HIV coreceptors ccr5 and cxcr4 protects CD4+ T cells from HIV-1 infection. Blood 2014, 123, 61–69. [Google Scholar] [CrossRef]
- Benjamin, R.; Berges, B.K.; Solis-Leal, A.; Igbinedion, O.; Strong, C.L.; Schiller, M.R. TALEN gene editing takes aim on HIV. Hum. Genet. 2016, 135, 1059–1070. [Google Scholar] [CrossRef]
- Magro, G.; Calistri, A.; Parolin, C. Targeting and Understanding HIV Latency: The CRISPR System against the Provirus. Pathogens 2021, 10, 1257. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.; Basukala, B.; Lee, S.; Gagne, M.; Wong, W.W.; Henderson, A.J. Targeted Chromatinization and Repression of HIV-1 Provirus Transcription with Repurposed CRISPR/Cas9. Viruses 2020, 12, 1154. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Das, A.T.; Berkhout, B. Tackling HIV Persistence: Pharmacological versus CRISPR-Based Shock Strategies. Viruses 2018, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.G.; Chung, C.-H.; Atkins, A.; Dampier, W.; Khalili, K.; Nonnemacher, M.R.; Wigdahl, B. Gene Editing of HIV-1 Co-receptors to Prevent and/or Cure Virus Infection. Front. Microbiol. 2018, 9, 2940. [Google Scholar] [CrossRef] [PubMed]
- Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Carette, J.E. A CRISPR toolbox to study virus-host interactions. Nat. Rev. Microbiol. 2017, 15, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Burdo, T.H.; Chen, C.; Kaminski, R.; Sariyer, I.K.; Mancuso, P.; Donadoni, M.; Smith, M.D.; Sariyer, R.; Caocci, M.; Liao, S.; et al. Preclinical safety and biodistribution of CRISPR targeting SIV in non-human primates. Gene Ther. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, C.; Zhang, T.; Li, F.; Yang, W.; Kaminski, R.; Fagan, P.R.; Putatunda, R.; Young, W.-B.; Khalili, K.; et al. CRISPR/gRNA-directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs. Sci. Rep. 2015, 5, 16277. [Google Scholar] [CrossRef]
- Saayman, S.M.; Lazar, D.C.; Scott, T.A.; Hart, J.R.; Takahashi, M.; Burnett, J.C.; Planelles, V.; Morris, K.V.; Weinberg, M.S. Potent and Targeted Activation of Latent HIV-1 Using the CRISPR/dCas9 Activator Complex. Mol. Ther. 2016, 24, 488–498. [Google Scholar] [CrossRef]
- Ji, H.; Jiang, Z.; Lu, P.; Ma, L.; Li, C.; Pan, H.; Fu, Z.; Qu, X.; Wang, P.; Deng, J.; et al. Specific Reactivation of Latent HIV-1 by dCas9-SunTag-VP64-mediated Guide RNA Targeting the HIV-1 Promoter. Mol. Ther. 2016, 24, 508–521. [Google Scholar] [CrossRef]
- Smith, L.M.; Hodara, V.L.; Parodi, L.M.; Callery, J.E.; Giavedoni, L.D. Silencing integrated SIV proviral DNA with TAR-specific CRISPR tools. J. Med. Primatol. 2020, 49, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Klinnert, S.; Chemnitzer, A.; Rusert, P.; Metzner, K.J. Systematic HIV-1 promoter targeting with CRISPR/dCas9-VPR reveals optimal region for activation of the latent provirus. J. Gen. Virol. 2022, 103, 1754. [Google Scholar] [CrossRef]
- da Costa, L.C.; Bomfim, L.M.; Dittz, U.V.T.; de Velozo, C.A.; da Cunha, R.D.; Tanuri, A. Repression of HIV-1 reactivation mediated by CRISPR/dCas9-KRAB in lymphoid and myeloid cell models. Retrovirology 2022, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Klinnert, S.; Schenkel, C.D.; Freitag, P.C.; Günthard, H.F.; Plückthun, A.; Metzner, K.J. Targeted shock-and-kill HIV-1 gene therapy approach combining CRISPR activation, suicide gene tBid and retargeted adenovirus delivery. Gene Ther. 2024, 31, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.A.; Chaudhry, W.; Campbell, L.A. Gesicles packaging dCas9-VPR ribonucleoprotein complexes can combine with vorinostat and promote HIV proviral transcription. Mol. Ther. Methods Clin. Dev. 2024, 32, 101203. [Google Scholar] [CrossRef] [PubMed]
- Bruggemans, A.; Vansant, G.; Balakrishnan, M.; Mitchell, M.L.; Cai, R.; Christ, F.; Debyser, Z. GS-9822, a preclinical LEDGIN candidate, displays a block-and-lock phenotype in cell culture. Antimicrob. Agents Chemother. 2023, 65, e02328-20. [Google Scholar] [CrossRef] [PubMed]
- Graves, P.; Zeng, Y. Biogenesis of mammalian microRNAs: A global view. Genom. Proteom. Bioinform. 2012, 10, 239–245. [Google Scholar] [CrossRef]
- Finnegan, E.F.; Pasquinelli, A.E. MicroRNA biogenesis: Regulating the regulators. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Chekulaeva, M.; Filipowicz, W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr. Opin. Cell Biol. 2009, 21, 452–460. [Google Scholar] [CrossRef]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef]
- Chinniah, R.; Adimulam, T.; Nandlal, L.; Arumugam, T.; Ramsuran, V. The Effect of miRNA Gene Regulation on HIV Disease. Front. Genet. 2022, 13, 862642. [Google Scholar] [CrossRef] [PubMed]
- Klase, Z.; Houzet, L.; Jeang, K.-T. MicroRNAs and HIV-1: Complex interactions. J. Biol. Chem. 2012, 287, 40884–40890. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.; Navas-Martín, S.; Martín-García, J. MicroRNAs and HIV-1 infection: Antiviral activities and beyond. J. Mol. Biol. 2014, 426, 1178–1197. [Google Scholar] [CrossRef] [PubMed]
- Nathans, R.; Chu, C.-Y.; Serquina, A.K.; Lu, C.-C.; Cao, H.; Rana, T.M. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol. Cell 2009, 34, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Sung, T.-L.; Rice, A.P. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009, 5, e1000263. [Google Scholar] [CrossRef]
- Chiang, K.; Sung, T.-L.; Rice, A.P. Regulation of cyclin T1 and HIV-1 Replication by microRNAs in resting CD4+ T lymphocytes. J. Virol. 2012, 86, 3244–3252. [Google Scholar] [CrossRef] [PubMed]
- Triboulet, R.; Mari, B.; Lin, Y.-L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.D.; Doms, R.W. Human immunodeficiency virus type 2. J. Gen. Virol. 2002, 83, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Koofhethile, C.K.; Gao, C.; Chang, C.; Lian, X.; Shapiro, R.; Yu, X.G.; Lichterfeld, M.; Kanki, P.J. The HIV-2 proviral landscape is dominated by defective proviruses. AIDS 2024, 38, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.; Vranckx, L.; Gijsbers, R.; Christ, F.; Debyser, Z. Insight into HIV-2 latency may disclose strategies for a cure for HIV-1 infection. J. Virus Erad. 2017, 3, 7–14. [Google Scholar] [CrossRef]
- Tong-Starksen, S.E.; Welsh, T.M.; Peterlin, B.M. Differences in transcriptional enhancers of HIV-1 and HIV-2. Response to T cell activation signals. J. Immunol. 1990, 145, 4348–4354. [Google Scholar] [CrossRef] [PubMed]
- Kas, K.; Voz, M.L.; Hensen, K.; Meyen, E.; Van de Ven, W.J. Transcriptional activation capacity of the novel PLAG family of zinc finger proteins. J. Biol. Chem. 1998, 273, 23026–23032. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Stallcup, M.R. Mouse Zac1, a transcriptional coactivator and repressor for nuclear receptors. Mol. Cell. Biol. 2000, 20, 1855–1867. [Google Scholar] [CrossRef]
- Huang, S.M.; Schönthal, A.H.; Stallcup, M.R. Enhancement of p53-dependent gene activation by the transcriptional coactivator Zac1. Oncogene 2001, 20, 2134–2143. [Google Scholar] [CrossRef]
- Vega-Benedetti, A.F.; Saucedo, C.; Zavattari, P.; Vanni, R.; Zugaza, J.L.; Parada, L.A. PLAGL1: An important player in diverse pathological processes. J. Appl. Genet. 2017, 58, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.D.; Telwatte, S.; Kumar, N.; Ferreira, F.; Martin, H.A.; Kadiyala, G.N.; Wedrychowski, A.; Moron-Lopez, S.; Chen, T.-H.; Goecker, E.A.; et al. Novel assays to investigate the mechanisms of latent infection with HIV-2. PLoS ONE 2022, 17, e0267402. [Google Scholar] [CrossRef]
- Wedrychowski, A.; Janssens, J.; Kim, S.J.; Kadiyala, G.N.; Telwatte, S.; Gaudreau, S.M.; Cu-Uvin, S.; Tsibris, A.; Yukl, S.A. HIV-2 Transcription in Blood CD4+ T-Cells Is Inhibited by Blocks to Elongation and Completion. In Proceedings of the Conference on Retroviruses and Opportunistic Infections (CROI) 2024, Denver, CO, USA, 3–6 March 2024. Poster 472. [Google Scholar]

{kind=link}
{kind=link}
| Compound | Family of Compounds | Type | Target | Clinical Status | References |
|---|---|---|---|---|---|
| Vorinostat | Histone deacetylase inhibitors (HDACi) | LRA | Histone deacetylase | FDA approved | [56,57,63,69,99] |
| Romidepsin | Histone deacetylase inhibitors (HDACi) | LRA | Histone deacetylase | FDA approved | [58,60,65,70,99] |
| Panobinostat | Histone deacetylase inhibitors (HDACi) | LRA | Histone deacetylase | FDA approved | [10,59,65,69,99] |
| Belinostat | Histone deacetylase inhibitors (HDACi) | LRA | Histone deacetylase | FDA approved | [99] |
| Apabetalone (RVX-208) | Bromodomain inhibitors (BETi) | LRA | BD2/BRD4 | Pre-clinical | [100,101,102] |
| CPI-203 | Bromodomain inhibitors (BETi) | LRA | BRD4 | Pre-clinical | [103] |
| I-BET-151 | Bromodomain inhibitors (BETi) | LRA | BD1, BD2/BRD4 | Pre-clinical | [62,104] |
| MMQO | Bromodomain inhibitors (BETi) | LRA | BRD2-4/BRDT | Pre-clinical | [105] |
| OTX-015 | Bromodomain inhibitors (BETi) | LRA | BRD2-4/BRDT | Pre-clinical | [106,107] |
| PFI-1 | Bromodomain inhibitors (BETi) | LRA | BRD2/BRD4 | Pre-clinical | [100] |
| UMB-136 | Bromodomain inhibitors (BETi) | LRA | BD1/BRD4 | Pre-clinical | [108] |
| Ciapavir | Small molecules mimetic of cIAP1 | LRA | cIAP1 | Pre-clinical | [67] |
| SBI-0637142; Debio-1143, AZD5582 | Small molecules mimetic of cIAP1 | LRA | cIAP1 | Pre-clinical | [76] |
| Nanoparticles containing Tat mRNA (T66) | Tat agonists | LRA | Tat | Pre-clinical | [73,74] |
| Compound | Family of Compounds | Type | Target | Clinical Status | References |
|---|---|---|---|---|---|
| didehydro-Cortistatin A (dCA) | Tat inhibitors | LPA | Tat | Pre-clinical | [11] |
| Dasatinib | Tyrosine kinase inhibitors (TKIs) | LPA | SAMHD1 | Pre-clinical | [109] |
| Manidipine hydrochloride | Noise suppressor of gene expression | LPA | Calcium channel blocker | Pre-clinical | [12] |
| TE-2, TE-10, TE-14, TE-20 | Thioredoxin reductase inhibitors | LPA | Thioredoxin reductase redox pathway | Pre-clinical | [110] |
| Senexin A | Cyclin-dependent protein kinases inhibitors (CDKi) | LPA | CDK8/CDK19 | Pre-clinical | [84] |
| Filgotinib | Janus kinase (JAK) inhibitor splicing modulator | LPA | JAK HIV mRNA | FDA-approved | [88] |
| Digoxin | Splicing modulator | LPA | HIV mRNA | FDA-approved | [87] |
| Leptomycin B | Nuclear export inhibitor | LPA | CRM1 | Pre-clinical | [91,93] |
| Benfluoron | Nuclear export inhibitor | LPA | Rev-RRE | Pre-clinical | [92] |
| Ivermectin | Nuclear export inhibitor | LPA | Importin B | Pre-clinical | [89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izquierdo-Pujol, J.; Puertas, M.C.; Martinez-Picado, J.; Morón-López, S. Targeting Viral Transcription for HIV Cure Strategies. Microorganisms 2024, 12, 752. https://doi.org/10.3390/microorganisms12040752
Izquierdo-Pujol J, Puertas MC, Martinez-Picado J, Morón-López S. Targeting Viral Transcription for HIV Cure Strategies. Microorganisms. 2024; 12(4):752. https://doi.org/10.3390/microorganisms12040752
Chicago/Turabian StyleIzquierdo-Pujol, Jon, Maria C. Puertas, Javier Martinez-Picado, and Sara Morón-López. 2024. "Targeting Viral Transcription for HIV Cure Strategies" Microorganisms 12, no. 4: 752. https://doi.org/10.3390/microorganisms12040752
APA StyleIzquierdo-Pujol, J., Puertas, M. C., Martinez-Picado, J., & Morón-López, S. (2024). Targeting Viral Transcription for HIV Cure Strategies. Microorganisms, 12(4), 752. https://doi.org/10.3390/microorganisms12040752