
Pannonibacter anstelovis sp. nov. Isolated from Two Cases of Bloodstream Infections in Paediatric Patients
 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. In Vitro Analysis
2.3. 16S rDNA Sanger Sequencing
2.4. Whole-Genome Sequencing
2.5. Bioinformatics Analyses
3. Results
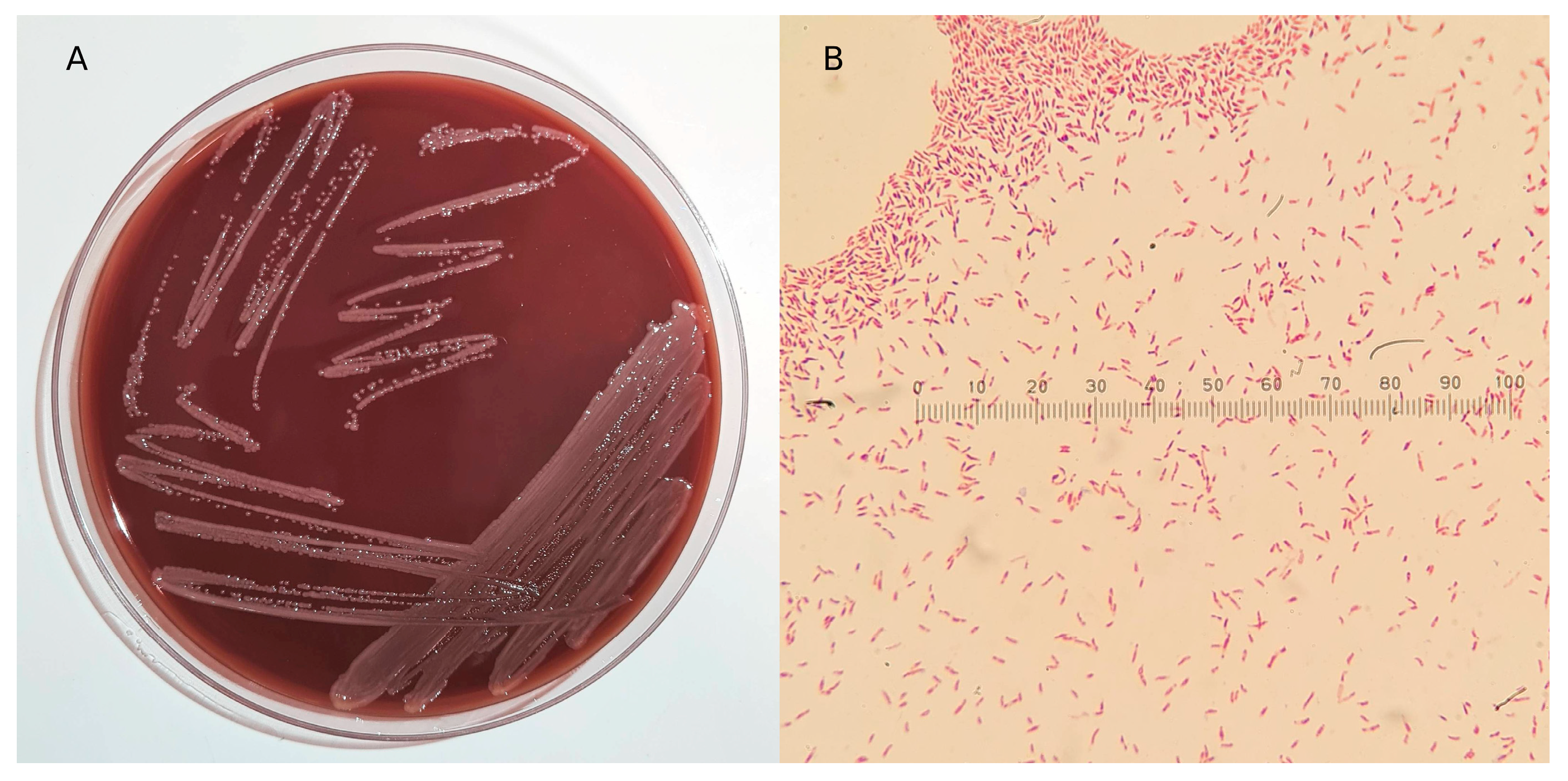
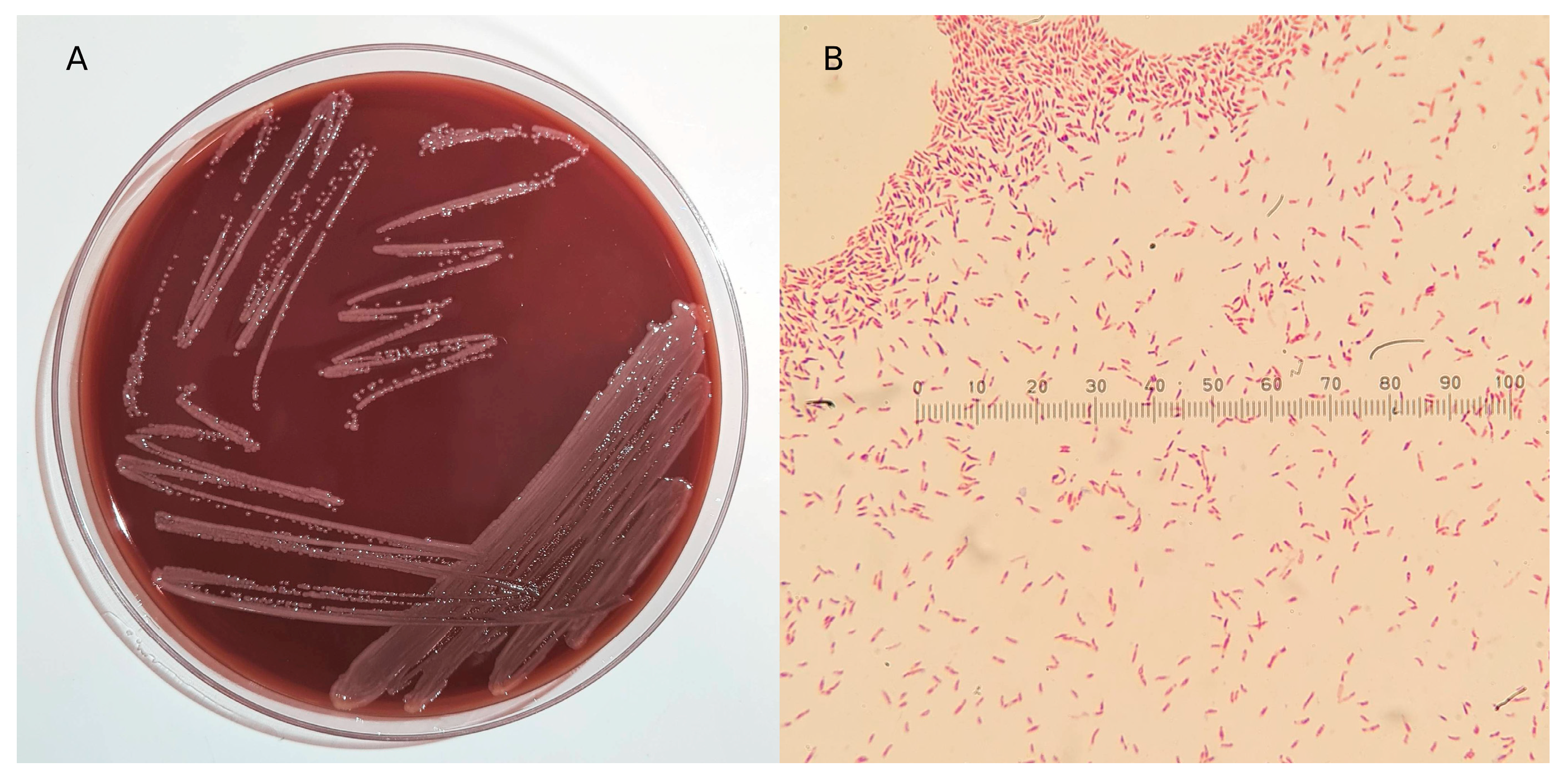
3.1. Isolation, Microscopic, and Biochemical Characterization
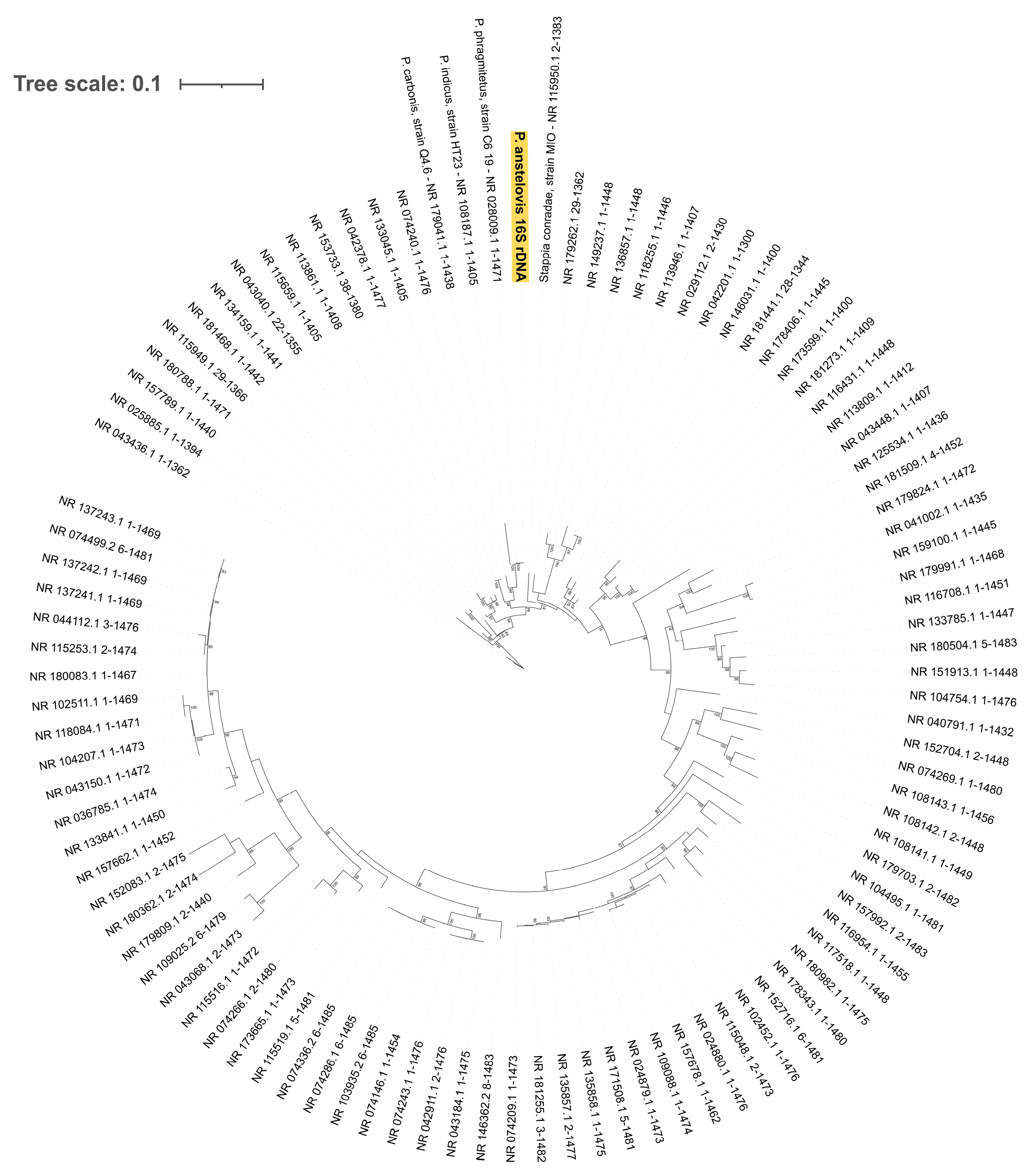
3.2. 16S Gene Analysis
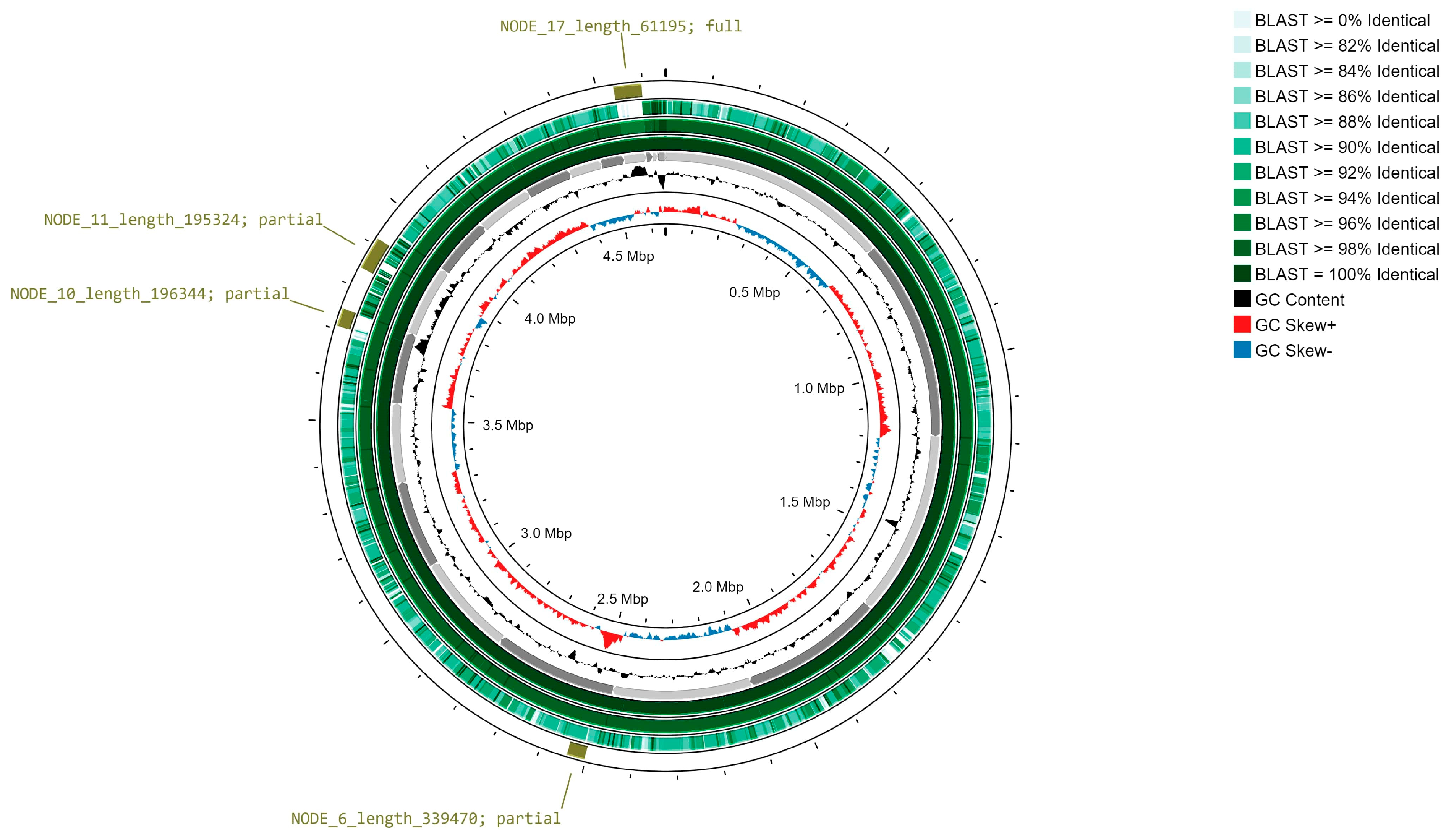
3.3. Paired-End Sequencing Analysis
3.4. Oxford Nanopore Technology Re-Sequencing Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xi, L.; Qiao, N.; Liu, D.; Li, J.; Zhang, J.; Liu, J. Pannonibacter carbonis sp. nov., isolated from coal mine water. Int. J. Syst. Evol. Microbiol. 2018, 68, 2042–2047. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Whitman, W.B.; Das, S.K. Draft Genome Sequence of Pannonibacter indicus Strain HT23 T (DSM 23407 T), a Highly Arsenate-Tolerant Bacterium Isolated from a Hot Spring in India. Genome Announc. 2017, 5, e00283-17. [Google Scholar] [CrossRef] [PubMed]
- Borsodi, A.K.; Micsinai, A.; Kovács, G.; Tóth, E.; Schumann, P.; Kovács, A.L.; Böddi, B.; Márialigeti, K. Pannonibacter phragmitetus gen. nov., sp. nov., a novel alkalitolerant bacterium isolated from decomposing reed rhizomes in a Hungarian soda lake. Int. J. Syst. Evol. Microbiol. 2003, 53, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, X.; Jiang, T.; Hu, S.; Yi, Z.; Zhou, Y.; Ming, D.; Chen, S. Liver Abscess Caused by Pannonibacter phragmitetus: Case Report and Literature Review. Front. Med. 2017, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, A.; Del Carmen Merino Bueno, M.; Merino, C.S.; Laurés, A.M.S.; De La Torre-Fernández, M.; Álvarez, E.S. First case of bac-teriemia caused by Pannonibacter phragmitetus in a haemodialysis patient. Nefrol. Engl. Ed. 2022, 42, 209–210. [Google Scholar]
- Tang, R.; Wang, J.; Zhan, Y.; Wu, K.; Wang, H.; Lu, Z. Hemodialysis catheter-related infection caused by Pannonibacter phragmitetus: A rare case report in China. Front. Cell. Infect. Microbiol. 2022, 12, 926154. [Google Scholar] [CrossRef] [PubMed]
- American Academy of Microbiology. From Outside to Inside: Environmental Microorganisms as Human Pathogens; American Society for Microbiology: Washington, DC, USA, 2004; Volume 16. [Google Scholar]
- Cerini, P.; Meduri, F.R.; Tomassetti, F.; Polidori, I.; Brugneti, M.; Nicolai, E.; Bernardini, S.; Pieri, M.; Broccolo, F. Trends in Antibiotic Resistance of Nosocomial and Community-Acquired Infections in Italy. Antibiotics 2023, 12, 651. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (U.S.). Antibiotic Resistance Threats in the United States. 2019. Available online: https://stacks.cdc.gov/view/cdc/82532 (accessed on 5 January 2024).
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilbäck, C.; Sandegren, L.; Hughes, D.; Andersson, D.I. Selection of Resistant Bacteria at Very Low Antibiotic Concentrations. PLoS Pathog. 2011, 7, e1002158. [Google Scholar] [CrossRef]
- Meier, H.; Spinner, K.; Crump, L.; Kuenzli, E.; Schuepbach, G.; Zinsstag, J. State of Knowledge on the Acquisition, Diversity, Inter-species Attribution and Spread of Antimicrobial Resistance between Humans, Animals and the Environment: A Systematic Review. Antibiotics 2022, 12, 73. [Google Scholar] [CrossRef]
- Winter, M.; Buckling, A.; Harms, K.; Johnsen, P.J.; Vos, M. Antimicrobial resistance acquisition via natural transformation: Context is everything. Curr. Opin. Microbiol. 2021, 64, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Bobate, S.; Mahalle, S.; Dafale, N.A.; Bajaj, A. Emergence of environmental antibiotic resistance: Mechanism, monitoring and management. Environ. Adv. 2023, 13, 100409. [Google Scholar] [CrossRef]
- Nossa, C.W. Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World J. Gastroenterol. 2010, 16, 4135. [Google Scholar] [CrossRef] [PubMed]
- Hall, T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/97/NT. In Nucleic Acids Symposium Series; Oxford University Press: Oxford, UK, 1999; pp. 95–98. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Capozzi, L.; Monno, M.R.; Del Sambro, L.; Manzulli, V.; Pesole, G.; Loconsole, D.; Parisi, A. Characterization of Bacillus cereus Group Isolates from Human Bacteremia by Whole-Genome Sequencing. Front. Microbiol. 2021, 11, 599524. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotech-nology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega. Curr. Protoc. Bioinform. 2014, 48, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.O.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable ge-nome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Teeling, H.; Meyerdierks, A.; Bauer, M.; Amann, R.; Glöckner, F.O. Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ. Microbiol. 2004, 6, 938–947. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.-A.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef]
- Seemann, T. ABRicate, Github 2020. Available online: https://github.com/tseemann/abricate (accessed on 22 May 2023).
- Knijn, A.; Michelacci, V.; Orsini, M.; Morabito, S. Advanced Research Infrastructure for Experimentation in GenomicS (ARIES): A lustrum of Galaxy Experience. Available online: http://biorxiv.org/lookup/doi/10.1101/2020.05.14.095901 (accessed on 22 May 2023).
- Carattoli, A.; Zankari, E.; Garcìa-Fernandez, A.; Larsen, M.; Lund, O.; Voldby Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef]
- The UniProt Consortium; Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies. MinKNOW. Oxford Science Park, OX4 4DQ, UK. 2023. Available online: https://community.nanoporetech.com/docs/prepare/library_prep_protocols/experiment-companion-minknow/v/mke_1013_v1_revcy_11apr2016 (accessed on 15 September 2023).
- De Coster, W.; Rademakers, R. NanoPack2: Population-scale evaluation of long-read sequencing data. Bioinformatics 2023, 39, btad311. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Oxford Nanopore Technologies Ltd. Medaka. Oxford Science Park, OX4 4DQ, UK. 2018. Available online: https://github.com/nanoporetech/medaka (accessed on 15 September 2023).
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.Y.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Starikova, E.V.; O Tikhonova, P.; A Prianichnikov, N.; Rands, C.M.; Zdobnov, E.M.; Ilina, E.N.; Govorun, V.M. Phigaro: High-throughput prophage sequence annotation. Bioinformatics 2020, 36, 3882–3884. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bliss, C.M.; Bennett, J.S.; Bratcher, H.B.; Brehony, C.; Colles, F.M.; Wimalarathna, H.; Harrison, O.B.; Sheppard, S.K.; Cody, A.J.; et al. Ribosomal multilocus sequence typing: Universal characterization of bacteria from domain to strain. Microbiology 2012, 158, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Wayne, L.G.; Moore, W.E.C.; Stackebrandt, E.; Kandler, O.; Colwell, R.R.; Krichevsky, M.I.; Truper, H.G.; Murray, R.G.E.; Grimont, P.A.D.; Brenner, D.J.; et al. Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int. J. Syst. Evol. Microbiol. 1987, 37, 463–464. [Google Scholar] [CrossRef]
- Church, D.L.; Cerutti, L.; Gürtler, A.; Griener, T.; Zelazny, A.; Emler, S. Performance and Application of 16S rRNA Gene Cycle Se-quencing for Routine Identification of Bacteria in the Clinical Microbiology Laboratory. Clin. Microbiol. Rev. 2020, 33, e00053-19. [Google Scholar] [CrossRef]
- Dohm, J.C.; Peters, P.; Stralis-Pavese, N.; Himmelbauer, H. Benchmarking of long-read correction methods. NAR Genom. Bioinform. 2020, 2, lqaa037. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Sahlin, K.; Baudeau, T.; Cazaux, B.; Marchet, C. A survey of mapping algorithms in the long-reads era. Genome Biol. 2023, 24, 133. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Molecule | MIC Pt 1 Strain | MIC Pt 2 Strain |
|---|---|---|
| Amikacin | >32 | >32 |
| Cefepime | 16 | 16 |
| Cefotaxime | 16 | 16 |
| Ceftazidime | 32 | 32 |
| Ciprofloxacin | 0.5 | 0.5 |
| Gentamycin | >8 | >8 |
| Imipenem | 0.5 | 0.5 |
| Meropenem | 1 | 1 |
| Piperacillin/Tazobactam | 16 | 16 |
| Tobramycin | >8 | >8 |
| Well | Biochemical Tests | Results |
|---|---|---|
| 0 | Control | − |
| 1 | Glycerol | − |
| 2 | Erythritol | − |
| 3 | D-Arabinose | W |
| 4 | L-Arabinose | + |
| 5 | D-Ribose | − |
| 6 | D-Xylose | + |
| 7 | L-Xylose | W |
| 8 | D-Adonitol | − |
| 9 | Methyl-βD-Xylopyranoside | − |
| 10 | D-Galactose | W |
| 11 | D-Glucose | W |
| 12 | D-Fructose | W |
| 13 | D-Mannose | − |
| 14 | L-Sorbose | − |
| 15 | L-Rhamnose | − |
| 16 | Dulcitol | − |
| 17 | Inositol | − |
| 18 | D-Mannitol | − |
| 19 | D-Sorbitol | − |
| 20 | Methyl-αD-Mannopyranoside | − |
| 21 | Methyl-αD-Glucopyranoside | − |
| 22 | N-AcetylGlucosamine | − |
| 23 | Amygdalin | − |
| 24 | Arbutin | − |
| 25 | Esculin ferric citrate | + |
| 26 | Salicin | − |
| 27 | D-Cellobiose | W |
| 28 | D-Maltose | − |
| 29 | D-Lactose | − |
| 30 | D-Melibiose | − |
| 31 | D-Saccharose (sucrose) | − |
| 32 | D-Trehalose | − |
| 33 | Inulin | − |
| 34 | D-Melezitose | − |
| 35 | D-Raffinose | − |
| 36 | Amidon (starch) | − |
| 37 | Glycogen | − |
| 38 | Xylitol | − |
| 39 | Gentiobiose | − |
| 40 | D-Turanose | − |
| 41 | D-Lyxose | − |
| 42 | D-Tagatose | − |
| 43 | D-fucose | + |
| 44 | L-fucose | W |
| 45 | D-Arabitol | − |
| 46 | L-Arabitol | − |
| 47 | Potassium Gluconate | − |
| 48 | Potassium 2-KetoGluconate | − |
| 49 | Potassium 5-KetoGluconate | − |
| Well | Biochemical Tests | Results |
|---|---|---|
| 1 | ONPG | + |
| 2 | ADH | − |
| 3 | LDC | − |
| 4 | ODC | − |
| 5 | Citrate | + |
| 6 | Hydrogen sulphide | − |
| 7 | Urease | − |
| 8 | TDA | − |
| 9 | Indole | − |
| 10 | Voges-Proskauer | − |
| 11 | Gelatin | − |
| 12 | Nitrate | − |
| Sample | Pt1 (MiSeq) | Pt2 (MiSeq) | Pt2 (ONT) | Pannonibacter phragmitetus DSM 14782 (GCA_000382365.1) |
|---|---|---|---|---|
| Pt1 (MiSeq) | - | 99.99 (99.71) | 99.76 (99.61) | 91.21 (85.76) |
| Pt2(MiSeq) | 100.00 (99.81) | - | 99.77 (99.65) | 91.23 (85.58) |
| Pt2-lr (ONT) | 99.78 (99.61) | 99.78 (99.67) | - | 91.16 (85.67) |
| Pannonibacter phragmitetus DSM 14782 (GCA_000382365.1) | 91.16 (83.33) | 91.16 (83.26) | 91.08 (83.29) | - |
| Protein Accession | InterPRO Retained Domain | Antimicrobial Putative Interaction |
|---|---|---|
| FGIJAEJN_03410 | Penicillin-binding protein, dimerisation domain | Cefepime, Cefotaxime, Ceftazidime, Piperacillin |
| FGIJAEJN_01305, FGIJAEJN_01921 | Penicillin-binding protein, transpeptidase | Cefepime, Cefotaxime, Ceftazidime, Piperacillin |
| FGIJAEJN_02309, FGIJAEJN_03733, FGIJAEJN_01441, FGIJAEJN_01366 | Beta-lactamase/transpeptidase-like | Cefepime, Cefotaxime, Ceftazidime, Piperacillin |
| FGIJAEJN_02847 | Penicillin-binding protein transglycosylase domain | Cefepime, Cefotaxime, Ceftazidime, Piperacillin |
| FGIJAEJN_00520 | Penicillin-binding protein, OB-like domain | Cefepime, Ceftazidime |
| FGIJAEJN_00222 | Penicillin-binding protein, C-terminal domain superfamily | Piperacillin |
| FGIJAEJN_03193, FGIJAEJN_01113, FGIJAEJN_03499, FGIJAEJN_02312, FGIJAEJN_02878, FGIJAEJN_00760 | Aminoglycoside phosphotransferase | Aminoglycoside |
| FGIJAEJN_00577, FGIJAEJN_02821, FGIJAEJN_02760, FGIJAEJN_03969, FGIJAEJN_00577, FGIJAEJN_03969 | Drug resistance transporter Bcr/CmlA subfamily | Bicyclomycin, Chloramphenicol, Florfenicol |
| FGIJAEJN_01136, FGIJAEJN_00166 | Chloramphenicol acetyltransferase-like domain superfamily | Chloramphenicol |
| FGIJAEJN_02702, FGIJAEJN_03166, FGIJAEJN_01924 | Glyoxalase/fosfomycin resistance/dioxygenase domain | Fosfomycin |
| FGIJAEJN_02566 | Multi antimicrobial extrusion protein | Multidrug |
| FGIJAEJN_00274 | Multiple antibiotic resistance (MarC)-related | Multidrug |
| FGIJAEJN_00230 | Drug resistance transporter EmrB-like | Multidrug |
| FGIJAEJN_00589 | Small drug/metabolite transporter protein family | Multidrug |
| FGIJAEJN_03037, FGIJAEJN_00275, FGIJAEJN_01585, FGIJAEJN_04086, FGIJAEJN_03816, FGIJAEJN_00630 | Multidrug efflux transporter AcrB TolC docking domain, DN/DC subdomains | Multidrug |
| FGIJAEJN_00589 | Small multidrug resistance protein | Multidrug |
| FGIJAEJN_04073 | Peptidase M74, penicillin-insensitive murein endopeptidase | Penicillin |
| FGIJAEJN_02114 | Penicillin-binding, C-terminal | Penicillin |
| FGIJAEJN_00838 | PBP domain | Penicillin |
| FGIJAEJN_02207 | Beta-lactamase-related | Penicillin, Cephamycin, Cephalosporin |
| FGIJAEJN_01279 | AmpG-like permease/Acetyl-coenzyme A transporter 1 | Penicillin, Cephamycin, Cephalosporin |
| FGIJAEJN_02140 | Beta-lactamase, class-A active site | Penicillin, Cephamycin, Cephalosporin |
| FGIJAEJN_00577 | Tetracycline resistance protein TetA/multidrug resistance protein MdtG | Tetracycline |
| FGIJAEJN_00237 | Tetracycline repressor TetR, C-terminal | Tetracycline |
| FGIJAEJN_04089 | Tetracyclin repressor-like, C-terminal domain | Tetracycline |
| FGIJAEJN_01776, FGIJAEJN_03091, FGIJAEJN_04181, FGIJAEJN_02679, FGIJAEJN_00245, FGIJAEJN_02722, FGIJAEJN_02364, FGIJAEJN_00123, FGIJAEJN_02082, FGIJAEJN_02903, FGIJAEJN_00246, FGIJAEJN_02749 | Tetracyclin repressor-like, C-terminal domain superfamily | Tetracycline |
| FGIJAEJN_03224 | PsrA, tetracyclin repressor-like, C-terminal domain | Tetracycline |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castellana, S.; De Laurentiis, V.; Bianco, A.; Del Sambro, L.; Grassi, M.; De Leonardis, F.; Derobertis, A.M.; De Carlo, C.; Sparapano, E.; Mosca, A.; et al. Pannonibacter anstelovis sp. nov. Isolated from Two Cases of Bloodstream Infections in Paediatric Patients. Microorganisms 2024, 12, 799. https://doi.org/10.3390/microorganisms12040799
Castellana S, De Laurentiis V, Bianco A, Del Sambro L, Grassi M, De Leonardis F, Derobertis AM, De Carlo C, Sparapano E, Mosca A, et al. Pannonibacter anstelovis sp. nov. Isolated from Two Cases of Bloodstream Infections in Paediatric Patients. Microorganisms. 2024; 12(4):799. https://doi.org/10.3390/microorganisms12040799
Chicago/Turabian StyleCastellana, Stefano, Vittoriana De Laurentiis, Angelica Bianco, Laura Del Sambro, Massimo Grassi, Francesco De Leonardis, Anna Maria Derobertis, Carmen De Carlo, Eleonora Sparapano, Adriana Mosca, and et al. 2024. "Pannonibacter anstelovis sp. nov. Isolated from Two Cases of Bloodstream Infections in Paediatric Patients" Microorganisms 12, no. 4: 799. https://doi.org/10.3390/microorganisms12040799