


Variations and Interseasonal Changes in the Gut Microbial Communities of Seven Wild Fish Species in a Natural Lake with Limited Water Exchange during the Closed Fishing Season


Abstract
1. Introduction
2. Materials and Methods
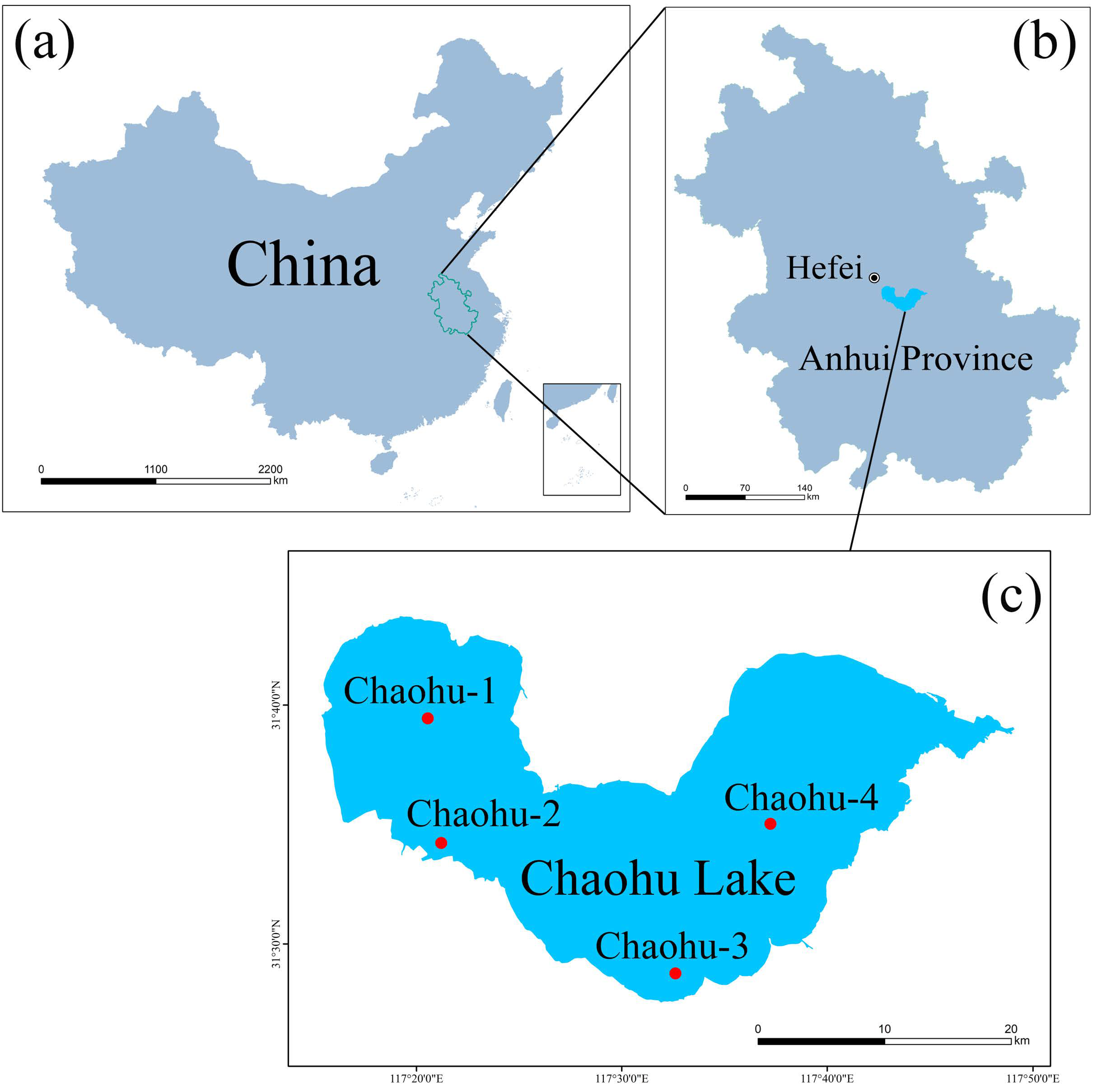
2.1. Sample Collection
2.2. DNA Extraction and Sequencing
2.3. Microbial Data Processing and Statistical Analysis
3. Results
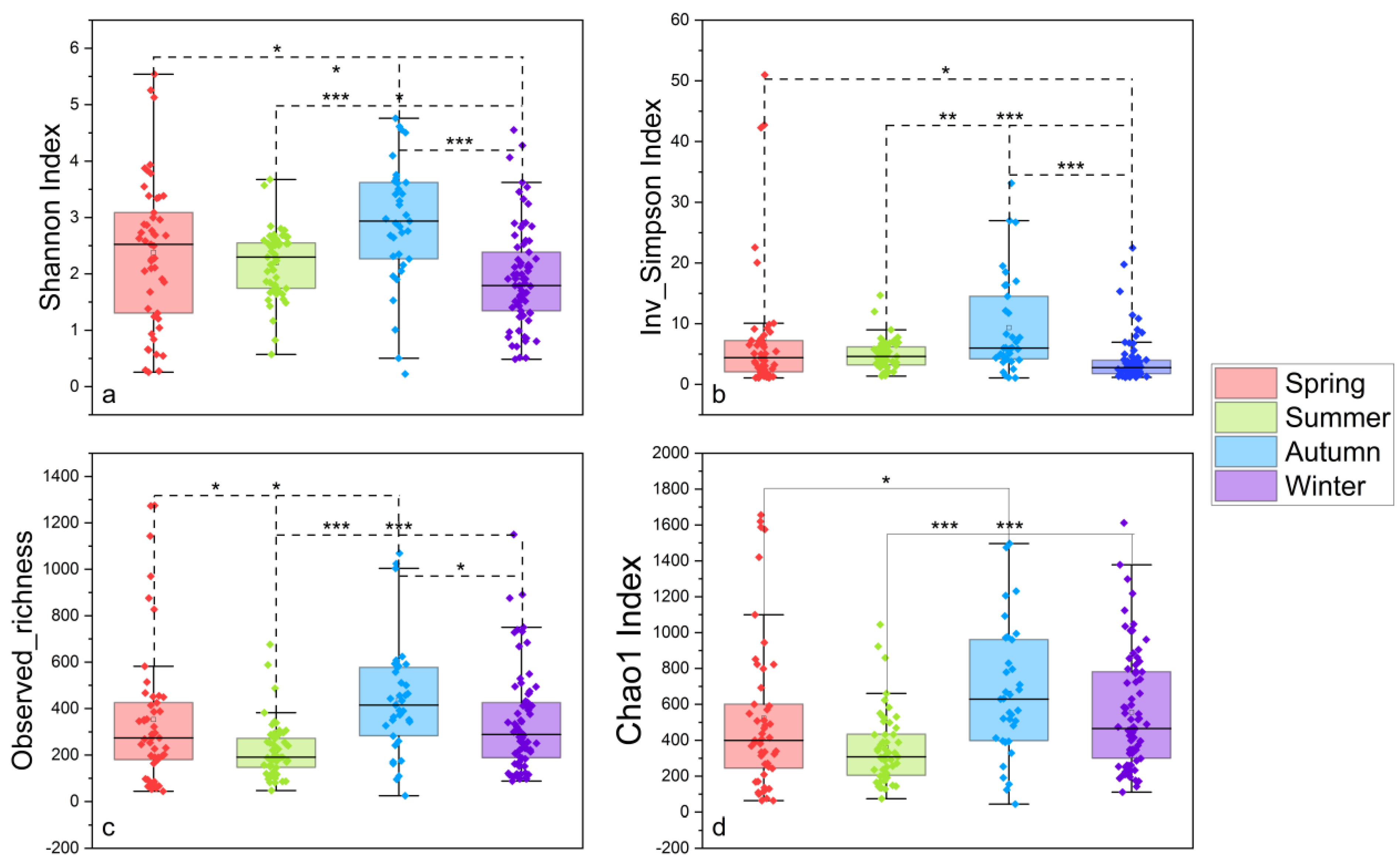
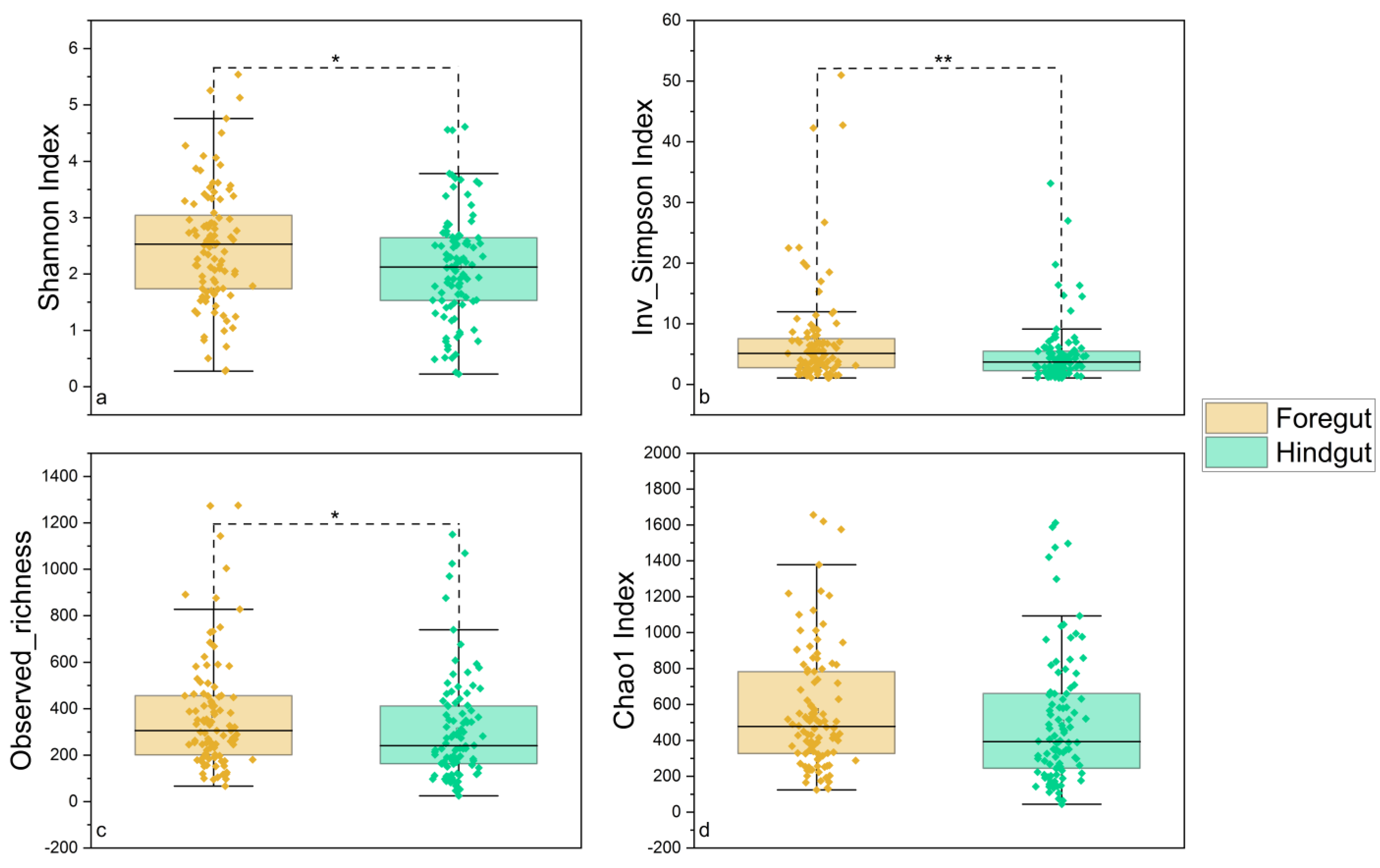
3.1. Sequencing Statistics and Microbial Diversity
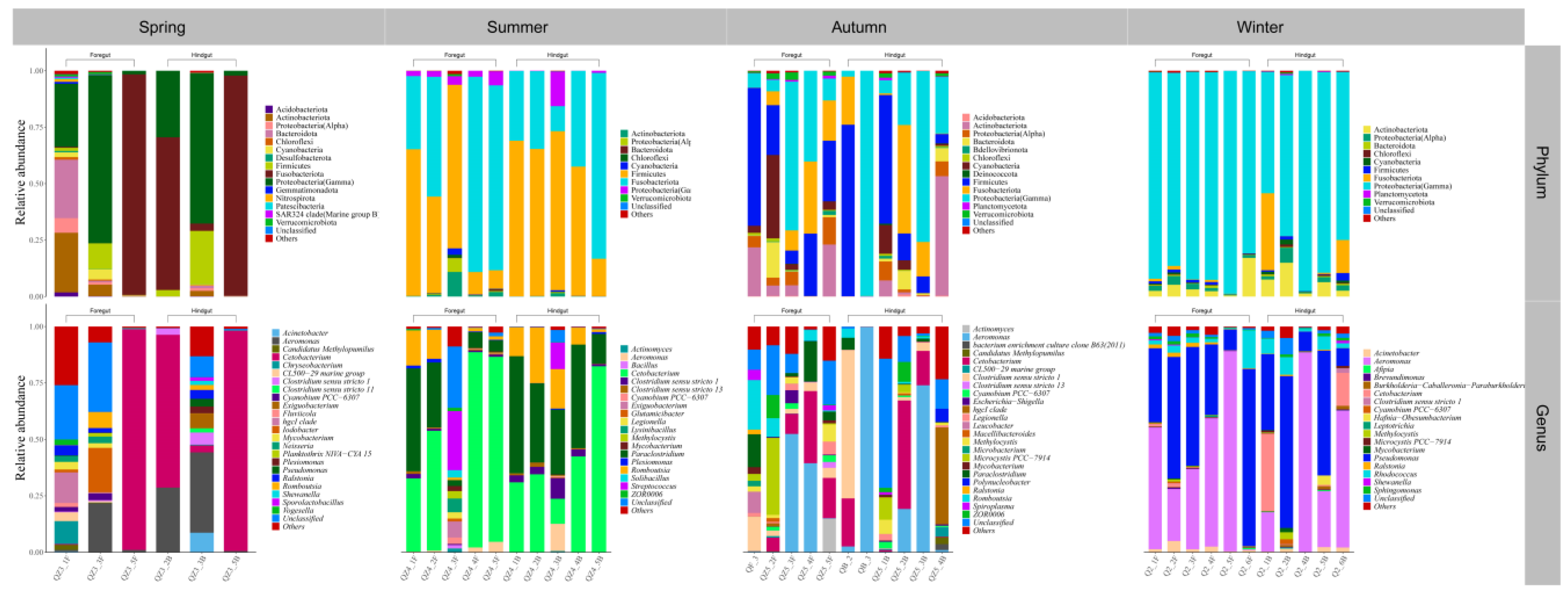
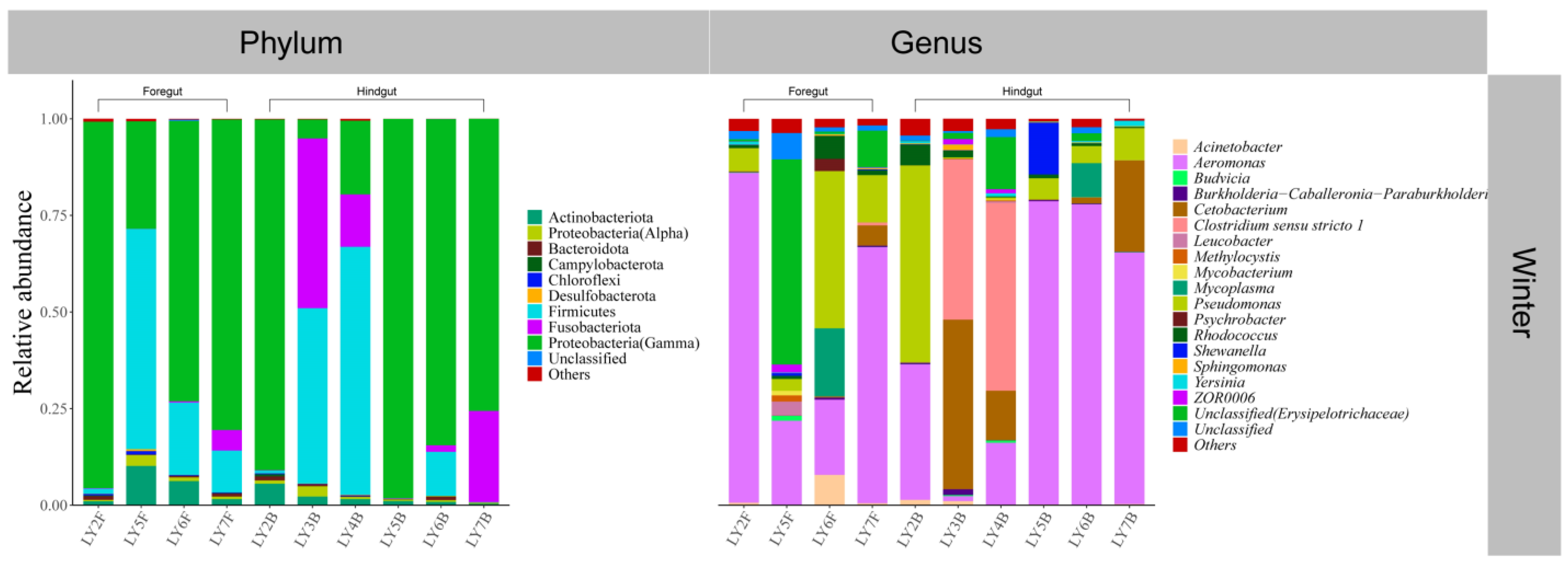
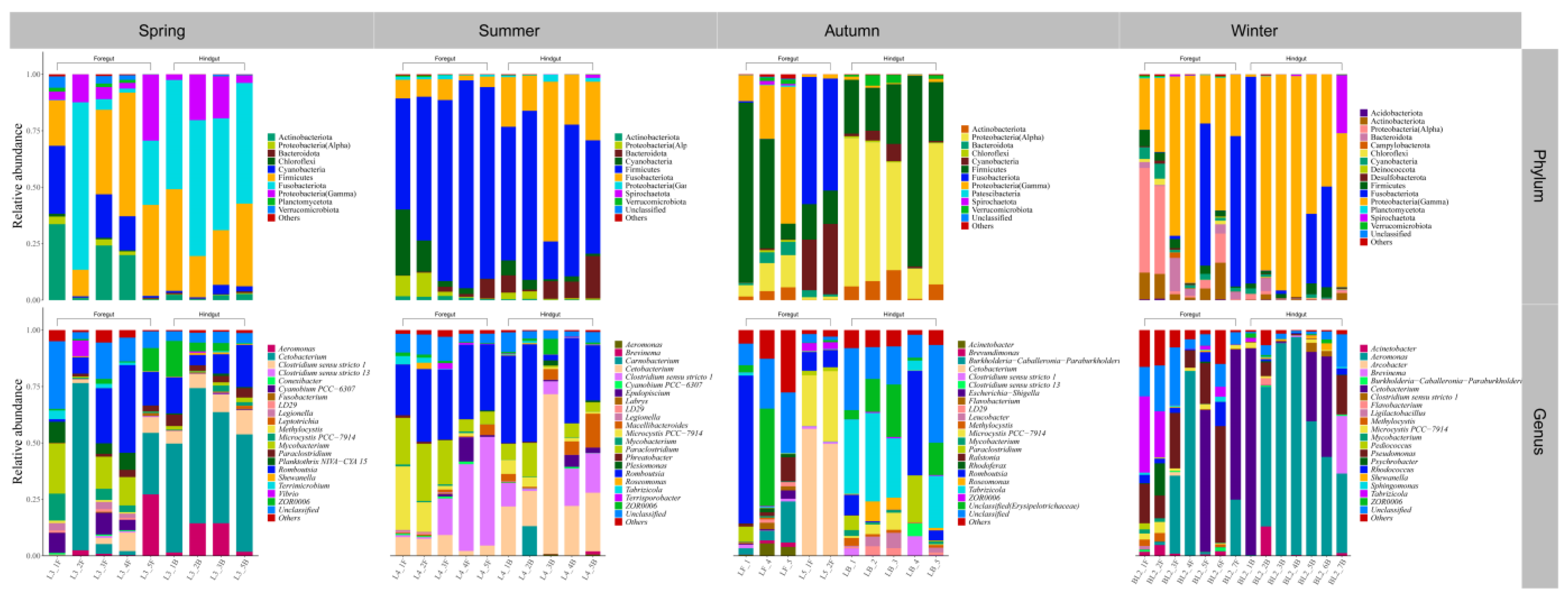
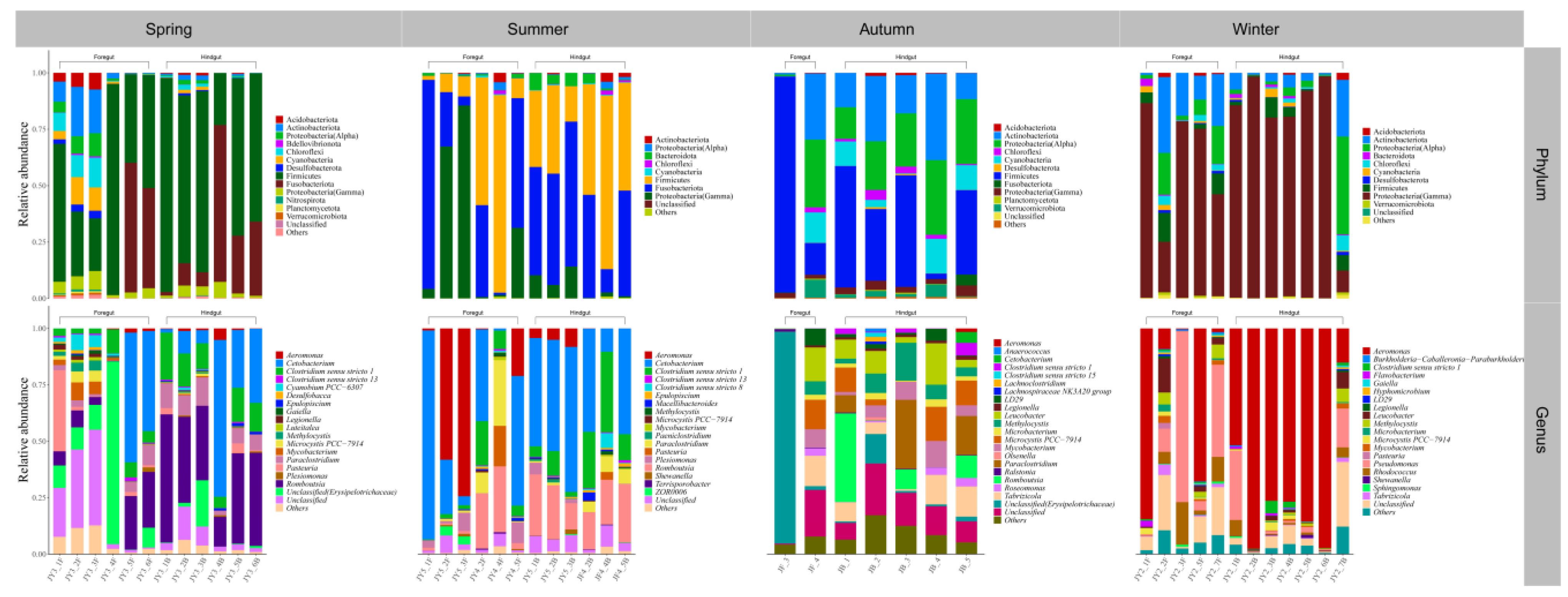
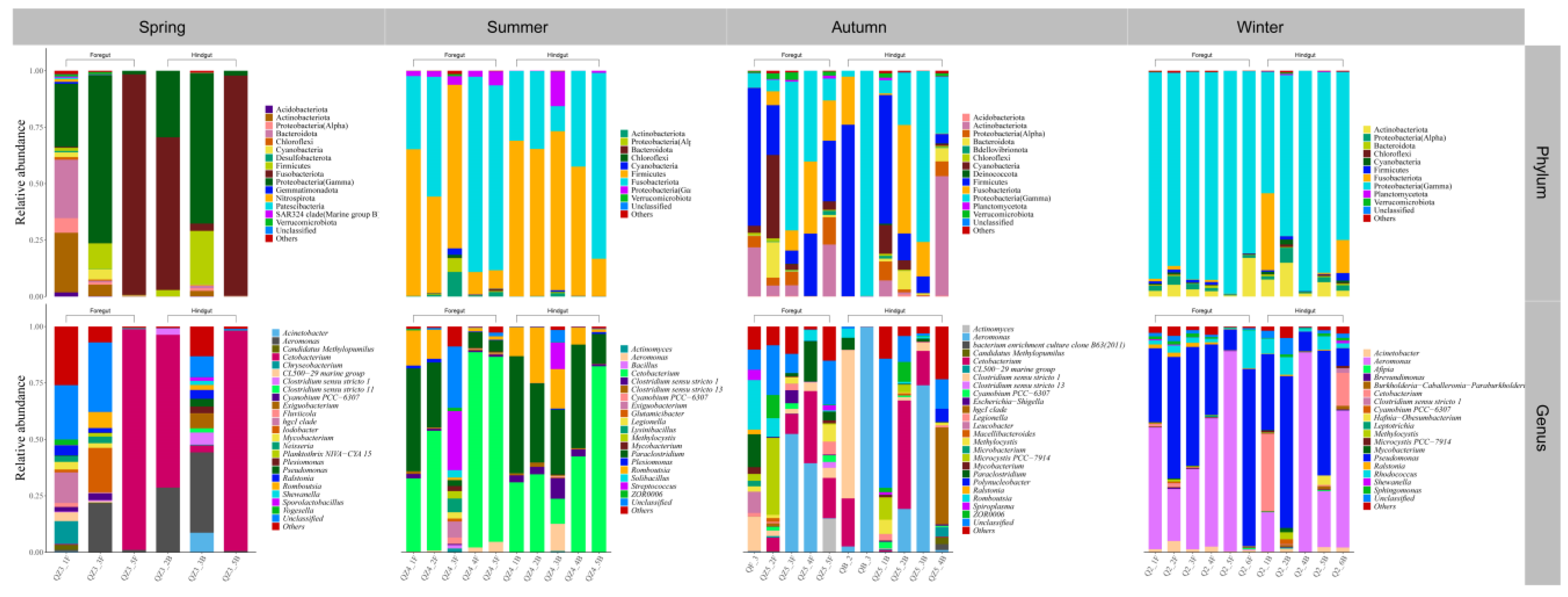
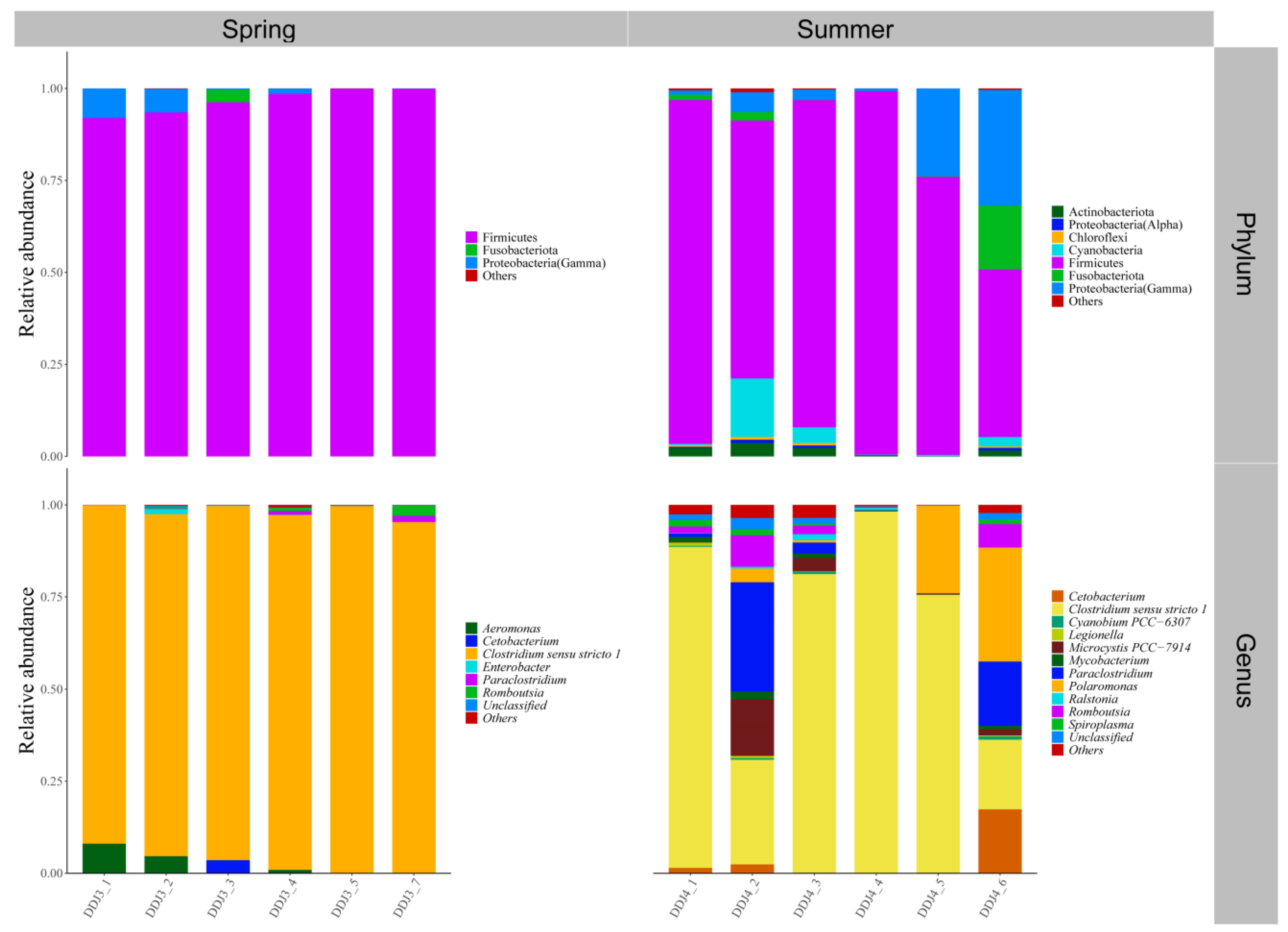
3.2. Gut Microbial Community Composition of Seven Wild Fish Species from Chaohu Lake
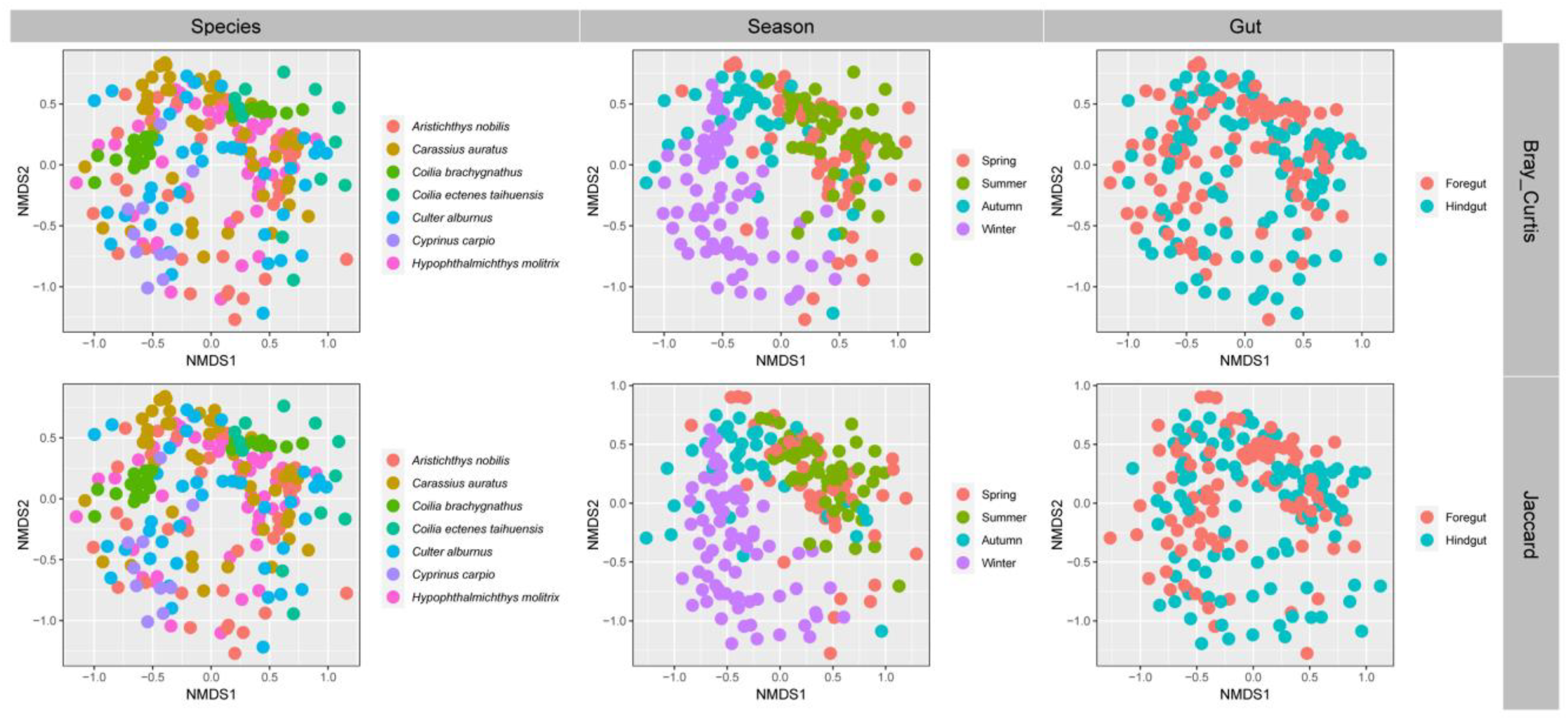
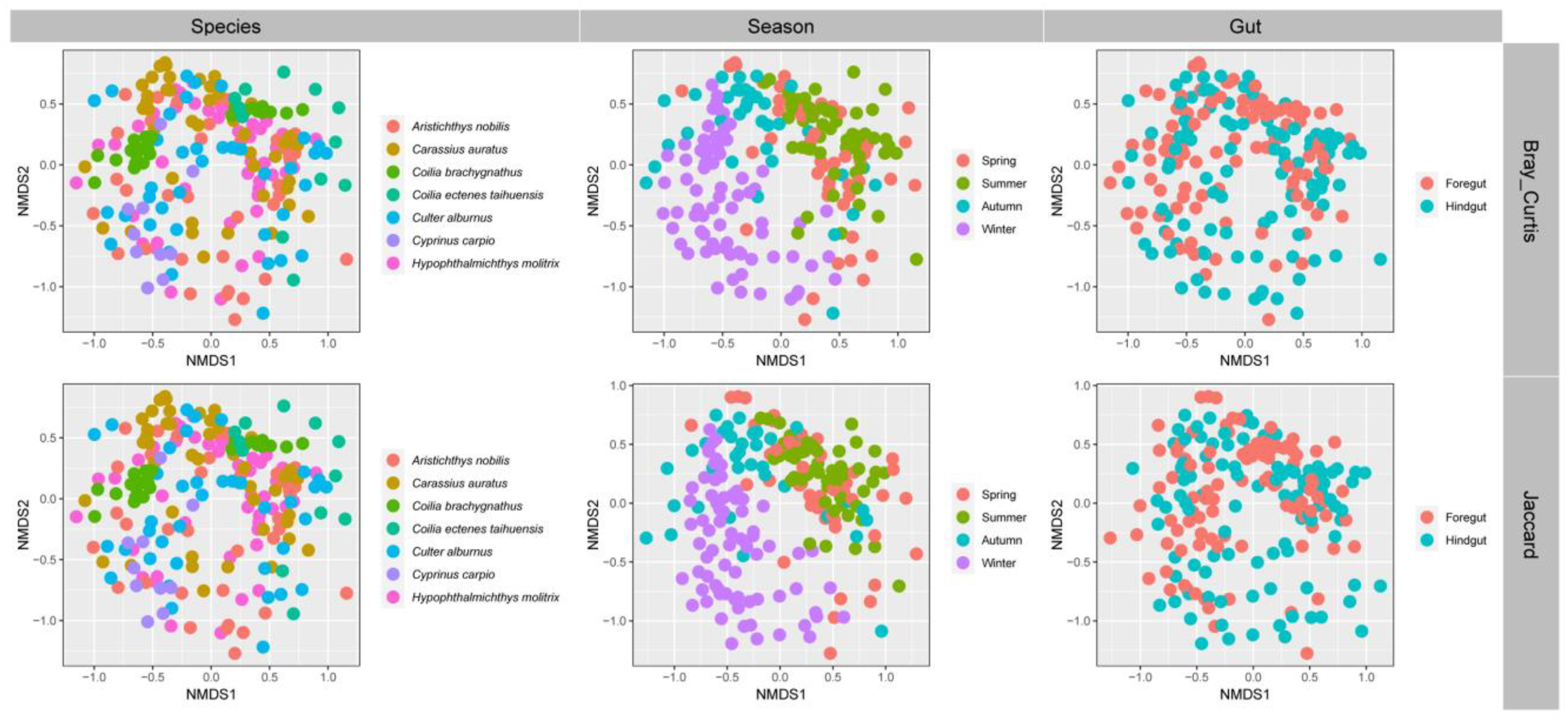
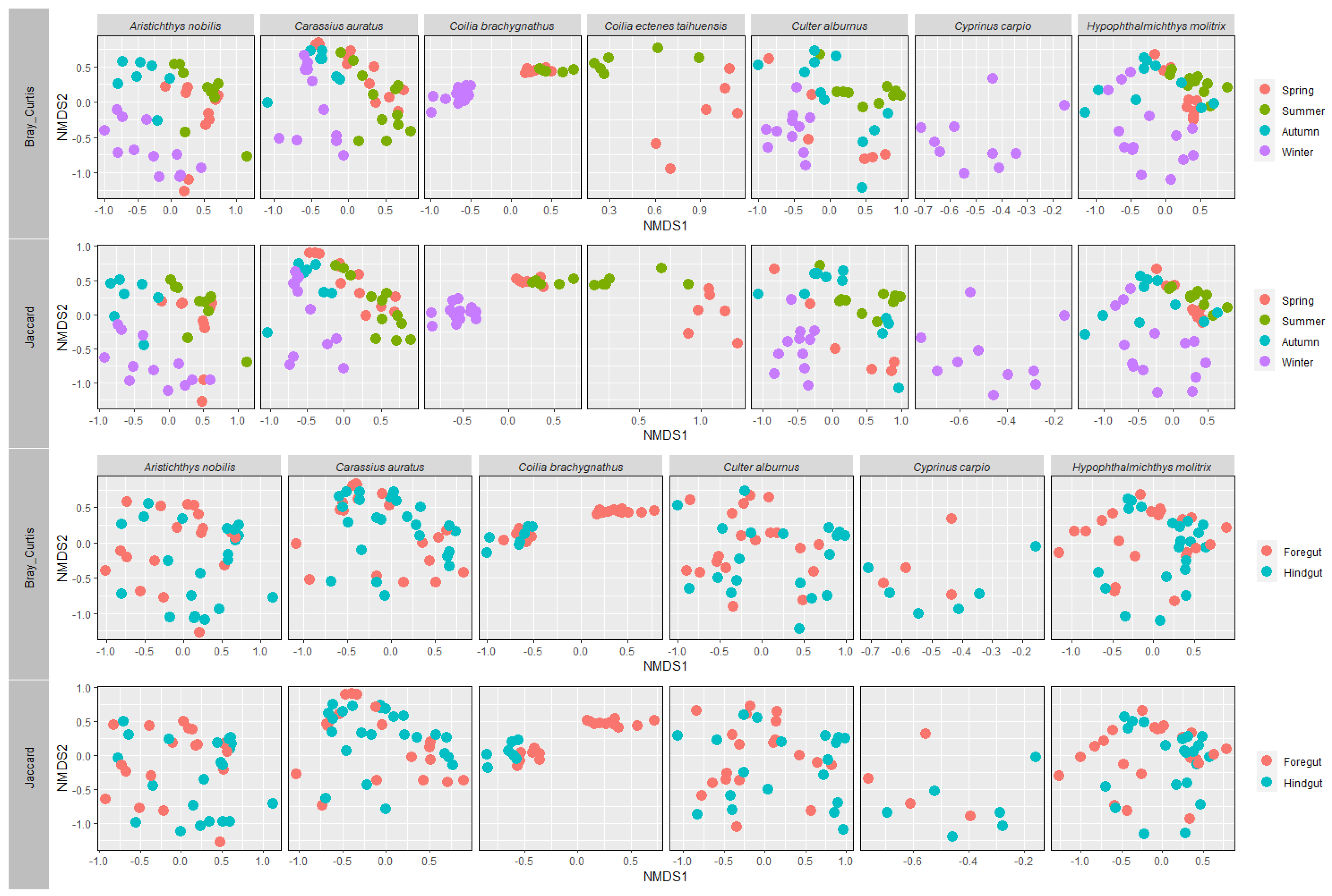
3.3. Gut Microbial Community Structure of Seven Wild Fish Species from Chaohu Lake
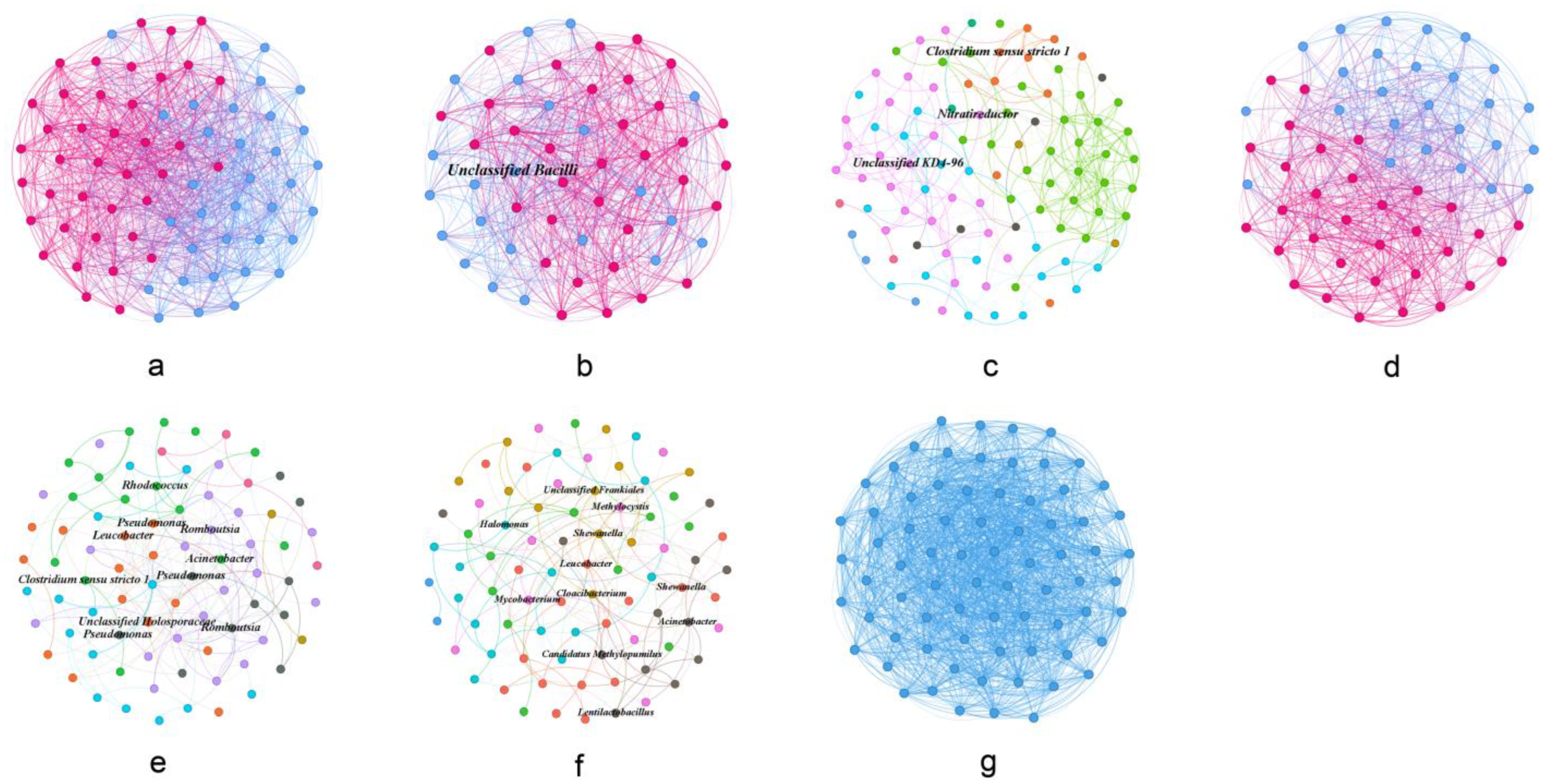
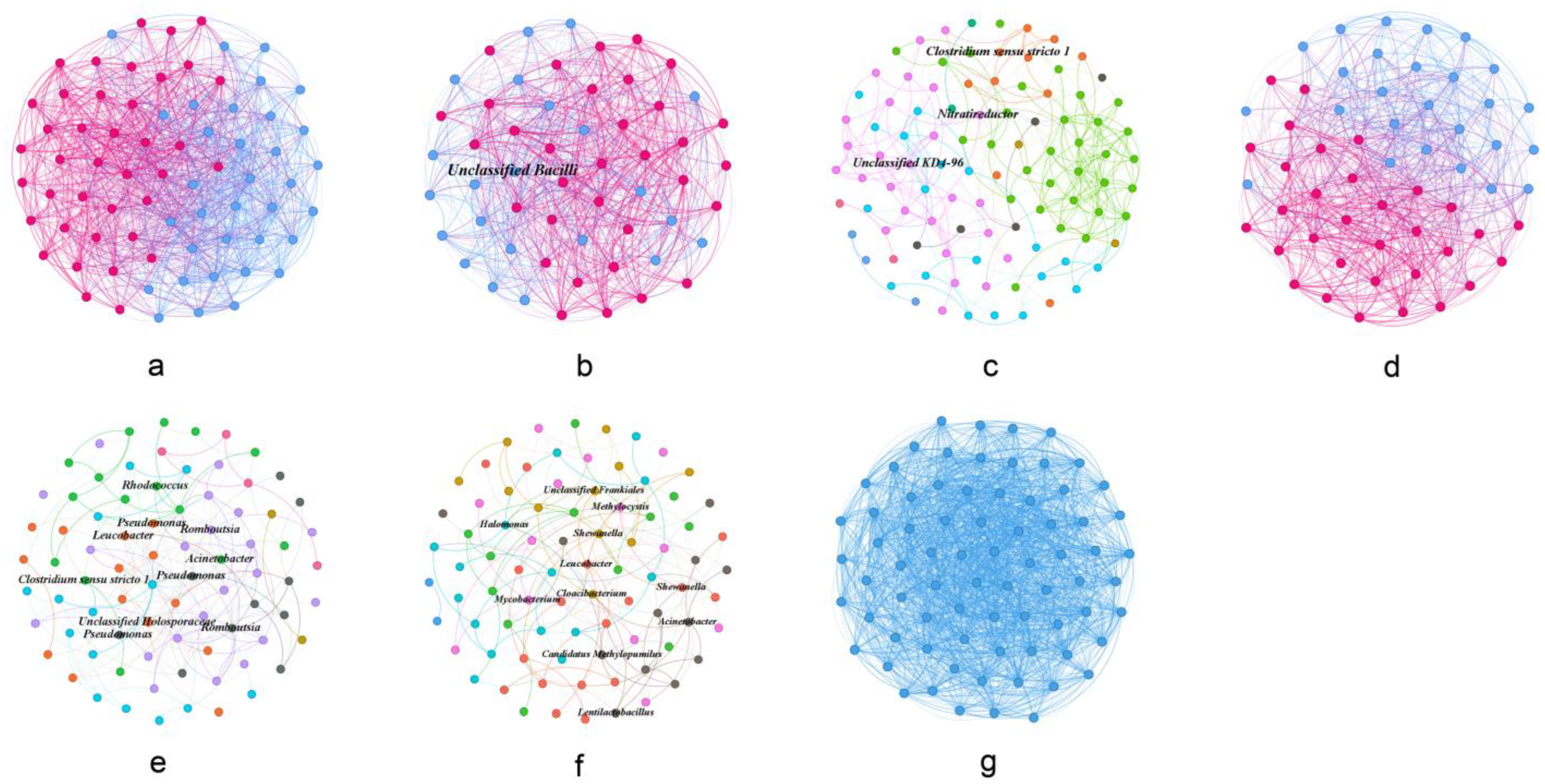
3.4. Co-Occurrence Network Profile of Gut Microbial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rawls, J.F.; Samuel, B.S.; Gordon, J.I. Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc. Natl. Acad. Sci. USA 2004, 101, 4596–4601. [Google Scholar] [CrossRef] [PubMed]
- Bates, J.M.; Mittge, E.; Kuhlman, J.; Baden, K.N.; Cheesman, S.E.; Guillemin, K. Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Dev. Biol. 2006, 297, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Rawls, J.F.; Mahowald, M.A.; Goodman, A.L.; Trent, C.M.; Gordon, J.I. In vivo imaging and genetic analysis link bacterial motility and symbiosis in the zebrafish gut. Proc. Natl. Acad. Sci. USA 2007, 104, 7622–7627. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.K. Role of gastrointestinal microbiota in fish. Aquac. Res. 2010, 41, 1553–1573. [Google Scholar] [CrossRef]
- Rhee, K.J.; Sethupathi, P.; Driks, A.; Lanning, D.K.; Knight, K.L. Role of commensal bacteria in development of gut-associated lymphoid tissues and preimmune antibody repertoire. J. Immunol. 2004, 172, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Ringø, E.; Jutfelt, F.; Kanapathippillai, P.; Bakken, Y.; Sundell, K.; Glette, J.; Mayhew, T.M.; Myklebust, R.; Olsen, R.E. Damaging effect of the fish pathogen Aeromonas salmonicida ssp. salmonicida on intestinal enterocytes of Atlantic salmon (Salmo salar L.) . Cell Tissue Res. 2004, 318, 305–311. [Google Scholar] [PubMed]
- Chen, Q.; Yan, Q.; Wang, K.; Zhuang, Z.; Wang, X. Portal of entry for pathogenic Vibrio alginolyticus into large yellow croaker Pseudosciaena crocea, and characteristics of bacterial adhesion to mucus. Dis. Aquat. Org. 2008, 80, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.S.; Grande, T.C.; Wilson, M.V.H. Fishes of the World; John Wiley and Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- BillaRd, R.; BeRni, P. Trends in cyprinid polyculture. Cybium 2004, 28, 255–261. [Google Scholar]
- Li, X.; Yu, Y.; Li, C.; Yan, Q. Comparative study on the gut microbiotas of four economically important Asian carp species. Sci. China Life Sci. 2018, 61, 696–705. [Google Scholar] [CrossRef]
- Shen, R.; Gu, X.; Chen, H.; Mao, Z.; Zeng, Q.; Jeppesen, E. Combining bivalve (Corbicula fluminea) and filter-feeding fish (Aristichthys nobilis) enhances the bioremediation effect of algae: An outdoor mesocosm study. Sci. Total Environ. 2020, 727, 138692. [Google Scholar] [CrossRef]
- Li, X.; Zhu, Y.; Ringø, E.; Wang, X.; Gong, J.; Yang, D. Intestinal microbiome and its potential functions in bighead carp (Aristichthys nobilis) under different feeding strategies. PeerJ 2018, 6, e6000. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wang, Z.; Wang, B.; Mei, H.; Zhai, X.; Zhuang, Z.; Chen, M.; Zhang, Y. Non-specific immunity associated gut microbiome in Aristichthys nobilis under different rearing strategies. Genes 2021, 12, 916. [Google Scholar] [CrossRef] [PubMed]
- Sula, E.; Aliko, V.; Barceló, D.; Faggio, C. Combined effects of moderate hypoxia, pesticides and PCBs upon crucian carp fish, Carassius carassius, from a freshwater lake-in situ ecophysiological approach. Aquat. Toxicol. 2020, 228, 105644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Lu, G.; Sun, Y.; Yan, Z.; Dang, T.; Liu, J. Metagenomic analysis explores the interaction of aged microplastics and roxithromycin on gut microbiota and antibiotic resistance genes of Carassius auratus. J. Hazard. Mater. 2022, 425, 127773. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, H.; Gatesoupe, F.J.; She, R.; Lin, Q.; Yan, X.; Li, J.; Li, X. Bacterial signatures of “Red-Operculum” disease in the gut of crucian carp (Carassius auratus). Microb. Ecol. 2017, 74, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Jeney, Z.; Jian, Z. Use and exchange of aquatic resources relevant for food and aquaculture: Common carp (Cyprinus carpio L.). Rev. Aquac. 2009, 1, 163–173. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Tan, H.; Yang, C.; Ren, L.; Liu, Q.; Wang, S.; Hu, F.; Xiao, J.; Zhao, R. Genetic effects on the gut microbiota assemblages of hybrid fish from parents with different feeding habits. Front. Microbiol. 2018, 9, 2972. [Google Scholar] [CrossRef]
- Yang, H.; Wu, J.; Du, H.; Zhang, H.; Li, J.; Wei, Q. Quantifying the colonization of environmental microbes in the fish gut: A case study of wild fish populations in the Yangtze River. Front. Microbiol. 2022, 12, 828409. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Xu, M.; Ying, C.; Yin, D.; Dai, P.; Yang, Y.; Ye, K.; Liu, K. The intestinal microbiota of lake anchovy varies according to sex, body size, and local habitat in Taihu Lake, China. MicrobiologyOpen 2020, 9, e00955. [Google Scholar] [CrossRef]
- Xiao, F.; Zhu, W.; Yu, Y.; Huang, J.; Li, J.; He, Z.; Wang, J.; Yin, H.; Yu, H.; Liu, S. Interactions and stability of gut microbiota in zebrafish increase with host development. Microbiol. Spectr. 2022, 10, e01696-01621. [Google Scholar] [CrossRef]
- Su, E.; Wu, Y.; Chen, P.; Yu, H.; Liu, S.; Luo, H.; Yang, Y.; Wang, C.; Shu, L.; Wu, B. Dietary selenium regulates the diversity and stability of microbial communities in stomach and intestine of rabbitfish (Siganus oramin). Aquaculture 2023, 563, 738979. [Google Scholar] [CrossRef]
- Dehler, C.E.; Secombes, C.J.; Martin, S.A. Environmental and physiological factors shape the gut microbiota of Atlantic salmon parr (Salmo salar L.). Aquaculture 2017, 467, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Snowberg, L.K.; Gregory Caporaso, J.; Knight, R.; Bolnick, D.I. Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. ISME J. 2015, 9, 2515–2526. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Long, M.; Li, H.; Gatesoupe, F.J.; Zhang, X.; Zhang, Q.; Feng, D.; Li, A. Multi-omics analysis reveals a correlation between the host phylogeny, gut microbiota and metabolite profiles in cyprinid fishes. Front. Microbiol. 2017, 8, 454. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Zhu, W.; Yu, Y.; He, Z.; Wu, B.; Wang, C.; Shu, L.; Li, X.; Yin, H.; Wang, J. Host development overwhelms environmental dispersal in governing the ecological succession of zebrafish gut microbiota. NPJ Biofilms Microbiomes 2021, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y. Btrim: A fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 2011, 98, 152–153. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016, 081257. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing. 2010. Available online: http://www.R-project.org/ (accessed on 9 January 2024).
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Jiang, Y.H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhang, P.; Qin, Y.; Tu, Q.; Yang, Y.; He, Z.; Schadt, C.W.; Zhou, J. Network succession reveals the importance of competition in response to emulsified vegetable oil amendment for uranium bioremediation. Environ. Microbiol. 2016, 18, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, M.S.; Boutin, S.; Hoseinifar, S.H.; Derome, N. Teleost microbiomes: The state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 2014, 5, 207. [Google Scholar] [CrossRef] [PubMed]
- Perry, W.B.; Lindsay, E.; Payne, C.J.; Brodie, C.; Kazlauskaite, R. The role of the gut microbiome in sustainable teleost aquaculture. Proc. R. Soc. B 2020, 287, 20200184. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, M.; Kneifel, W.; Domig, K.J. A new view of the fish gut microbiome: Advances from next-generation sequencing. Aquaculture 2015, 448, 464–475. [Google Scholar] [CrossRef]
- Luna, G.M.; Quero, G.M.; Kokou, F.; Kormas, K. Time to integrate biotechnological approaches into fish gut microbiome research. Curr. Opin. Biotechnol. 2022, 73, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.R.; Ran, C.; Ringø, E.; Zhou, Z.G. Progress in fish gastrointestinal microbiota research. Rev. Aquac. 2018, 10, 626–640. [Google Scholar] [CrossRef]
- Pessoa, R.B.G.; de Oliveira, W.F.; Marques, D.S.C.; dos Santos Correia, M.T.; de Carvalho, E.V.M.M.; Coelho, L.C.B.B. The genus Aeromonas: A general approach. Microb. Pathog. 2019, 130, 81–94. [Google Scholar] [CrossRef]
- Dallaire-Dufresne, S.; Tanaka, K.H.; Trudel, M.V.; Lafaille, A.; Charette, S.J. Virulence, genomic features, and plasticity of Aeromonas salmonicida subsp. salmonicida, the causative agent of fish furunculosis. Vet. Microbiol. 2014, 169, 1–7. [Google Scholar]
- Zhao, Y.; Li, S.; Lessing, D.J.; Guo, L.; Chu, W. Characterization of Cetobacterium somerae CPU-CS01 isolated from the intestine of healthy crucian carp (Carassius auratus) as potential probiotics against Aeromonas hydrophila infection. Microb. Pathog. 2023, 180, 106148. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Xie, Y.; Li, Y.; Zhou, W.; Zhang, Z.; Yang, Y.; Olsen, R.E.; Ringø, E.; Ran, C.; Zhou, Z. Stabilized fermentation product of Cetobacterium somerae improves gut and liver health and antiviral immunity of zebrafish. Fish Shellfish Immunol. 2022, 120, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhang, Z.; Ding, Q.; Yang, Y.; Bindelle, J.; Ran, C.; Zhou, Z. Intestinal Cetobacterium and acetate modify glucose homeostasis via parasympathetic activation in zebrafish. Gut Microbes 2021, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Zhang, P.; Lin, M.; Lin, W.; Yang, Z.; Li, S. Microbial diversity and structure in the gastrointestinal tracts of two stranded short-finned pilot whales (Globicephala macrorhynchus) and a pygmy sperm whale (Kogia breviceps). Integr. Zool. 2021, 16, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Zhang, P.; Zhang, C.; Du, J.; Du, X.; Zhu, C.; Liu, J.; Xie, P.; Li, S. Comparative study of the gut microbiota among four different marine mammals in an aquarium. Front. Microbiol. 2021, 12, 769012. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Zhang, P.; Zhang, X.; Yang, Z.; Li, S. Gut microbial characterization of melon-headed whales (Peponocephala electra) stranded in China. Microorganisms 2022, 10, 572. [Google Scholar] [CrossRef] [PubMed]
- Marón, C.F.; Kohl, K.D.; Chirife, A.; Di Martino, M.; Fons, M.P.; Navarro, M.A.; Beingesser, J.; McAloose, D.; Uzal, F.A.; Dearing, M.D. Symbiotic microbes and potential pathogens in the intestine of dead southern right whale (Eubalaena australis) calves. Anaerobe 2019, 57, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Li, H.; Ma, R.; Ren, M.; Li, Y.; Li, J.; Chen, H.; Chen, Z.; Gong, D.; Wang, J. Effect of Coptis chinensis franch and Magnolia officinalis on intestinal flora and intestinal barrier in a TNBS-induced ulcerative colitis rats model. Phytomedicine 2022, 97, 153927. [Google Scholar] [CrossRef] [PubMed]
- Ricaboni, D.; Mailhe, M.; Khelaifia, S.; Raoult, D.; Million, M. Romboutsia timonensis, a new species isolated from human gut. New Microbes New Infect. 2016, 12, 6–7. [Google Scholar] [CrossRef]
- Chang, S.C.; Kao, M.R.; Saldivar, R.K.; Díaz-Moreno, S.M.; Xing, X.; Furlanetto, V.; Yayo, J.; Divne, C.; Vilaplana, F.; Abbott, D.W. The Gram-positive bacterium Romboutsia ilealis harbors a polysaccharide synthase that can produce (1, 3; 1, 4)-β-D-glucans. Nat. Commun. 2023, 14, 4526. [Google Scholar] [CrossRef]
- Duman, M.; Mulet, M.; Altun, S.; Saticioglu, I.B.; Ozdemir, B.; Ajmi, N.; Lalucat, J.; García-Valdés, E. The diversity of Pseudomonas species isolated from fish farms in Turkey. Aquaculture 2021, 535, 736369. [Google Scholar] [CrossRef]
- Kho, C.J.Y.; Lau, M.M.L.; Chung, H.H.; Chew, I.Y.Y.; Gan, H.M. Whole-genome sequencing of Pseudomonas koreensis isolated from diseased Tor tambroides. Curr. Microbiol. 2023, 80, 255. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Tu, Q.; Zhi, X. Functional molecular ecological networks. MBio 2010, 1, e00169-10. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Yang, Y. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. MBio 2011, 2, e00122-11. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Spring | Summer | Autumn | Winter | |
|---|---|---|---|---|---|
| Silver carp (Hypophthalmichthys molitrix) | Foregut | L3-1F, L3-2F, L3-3F, L3-4F, L3-5F | L4-1F, L4-2F, L4-3F, L4-4F, L4-5F | LF-1, LF4, LF5, L5-1F, L5-2F | BL2-1F, BL2-2F, BL2-3F, BL2-4F, BL2-5F, BL2-6F, BL2-7F |
| Hindgut | L3-1B, L3-2B, L3-3B, L3-5B | L4-1B, L4-2B, L4-3B, L4-4B, L4-5B | LB1, LB2, LB3, LB4, LB5 | BL2-1B, BL2-2B, BL2-3B, BL2-4B, BL2-5B, BL2-6B, BL2-7B | |
| Bighead carp (Aristichthys nobilis) | Foregut | Y3-1F, Y3-2F, Y3-3F, Y3-4F, Y3-5F | Y4-1F, Y4-2F, Y4-3F, Y4-4F, Y4-5F | YF-2, YF-3 | HL2-1F, HL2-3F, HL2-4F, HL2-5F, HL2-6F, HL2-7F |
| Hindgut | Y3-1B, Y3-2B, Y3-3B, Y3-4B, Y3-5B | Y4-1B, Y4-2B, Y4-3B, Y4-4B, Y4-5B | YB-1, YB-2, YB-3, YB-4, YB-5 | HL2-1B, HL2-2B, HL2-3B, HL2-4B, HL2-5B, HL2-6B | |
| Crucian carp (Carassius auratus) | Foregut | JY3-1F, JY3-2F, JY3-3F, JY3-4F, JY3-5F, JY3-6F | JY5-1F, JY5-2F, JY5-3F, JY4-2F, JY4-4F, JY4-5F | JF-3, JF-4 | JY2-1F, JY2-2F, JY2-3F, JY2-5F, JY2-7F |
| Hindgut | JY3-1B, JY3-2B, JY3-3B, JY3-4B, JY3-5B, JY3-6B | JY5-1B, JY5-2B, JY5-3B, JF4-2B, JF4-4B, JF4-5B | JB-1, JB-2, JB-3, JB-4, JB-5 | JY2-1B, JY2-2B, JY2-3B, JY2-4B, JY2-5B, JY2-6B, JY2-7B | |
| Topmouth culter (Culter alburnus) | Foregut | QZ3-1F, QZ3-3F, QZ3-5F | QZ4-1F, QZ4-2F, QZ4-3F, QZ4-4F, QZ4-5F | QF-3, QZ5-2F, QZ5-3F, QZ5-4F, QZ5-5F | Q2-1F, Q2-2F, Q2-3F, Q2-4F, Q2-5F, Q2-6F |
| Hindgut | QZ3-2B, QZ3-3B, QZ3-5B | QZ4-1B, QZ4-2B, QZ4-3B, QZ4-4B, QZ4-5B | QB-2, QB-3, QZ5-1B, QZ5-2B, QZ5-3B, QZ5-4B | Q2-1B, Q2-2B, Q2-4B, Q2-5B, Q2-6B | |
| Coilia brachygnathus | Foregut | DJ3-1F, DJ3-2F, DJ3-3F, DJ3-4F, DJ3-5F, DJ3-6F | XDJ4-1, XDJ4-2, XDJ4-3, XDJ4-4, XDJ4-5 | DJ1F, DJ2F, DJ3F, DJ4F, DJ5F, DJ6F, DJ7F | |
| Hindgut | DJ1B, DJ2B, DJ3B, DJ4B, DJ5B, DJ6B, DJ7B | ||||
| Common carp (Cyprinus carpio) | Foregut | LY2F, LY5F, LY6F, LY7F | |||
| Hindgut | LY2B, LY3B, LY4B, LY5B, LY6B, LY7B | ||||
| Lake anchovy (Coilia ectenes taihuensis) | DDJ3-1, DDJ3-2, DDJ3-3, DDJ3-4, DDJ3-5, DDJ3-7 | DDJ4-1, DDJ4-2, DDJ4-3, DDJ4-4, DDJ4-5, DDJ4-6 |
| Species | Node | Link | Average Clustering Coefficient (avgCC) | Average Path Distance (GD) | Modularity (M) |
|---|---|---|---|---|---|
| H. molitrix | 73 | 1151 | 0.532 | 1.563 | 0.117 |
| A. nobilis | 55 | 743 | 0.565 | 1.500 | 0.078 |
| C. auratus | 100 | 200 | 0.268 | 4.116 | 0.535 |
| C. alburnus | 58 | 678 | 0.465 | 1.592 | 0.133 |
| C. brachygnathus | 80 | 185 | 0.127 | 3.103 | 0.438 |
| C. carpio | 82 | 142 | 0.039 | 3.488 | 0.494 |
| C. ectenes taihuensis | 74 | 1623 | 0.628 | 1.399 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.; Wang, Z.; Gao, N.; Qi, X.; Zeng, J.; Cui, K.; Lu, W.; Bai, S. Variations and Interseasonal Changes in the Gut Microbial Communities of Seven Wild Fish Species in a Natural Lake with Limited Water Exchange during the Closed Fishing Season. Microorganisms 2024, 12, 800. https://doi.org/10.3390/microorganisms12040800
Liang Y, Wang Z, Gao N, Qi X, Zeng J, Cui K, Lu W, Bai S. Variations and Interseasonal Changes in the Gut Microbial Communities of Seven Wild Fish Species in a Natural Lake with Limited Water Exchange during the Closed Fishing Season. Microorganisms. 2024; 12(4):800. https://doi.org/10.3390/microorganisms12040800
Chicago/Turabian StyleLiang, Yangyang, Zijia Wang, Na Gao, Xiaoxue Qi, Juntao Zeng, Kai Cui, Wenxuan Lu, and Shijie Bai. 2024. "Variations and Interseasonal Changes in the Gut Microbial Communities of Seven Wild Fish Species in a Natural Lake with Limited Water Exchange during the Closed Fishing Season" Microorganisms 12, no. 4: 800. https://doi.org/10.3390/microorganisms12040800
APA StyleLiang, Y., Wang, Z., Gao, N., Qi, X., Zeng, J., Cui, K., Lu, W., & Bai, S. (2024). Variations and Interseasonal Changes in the Gut Microbial Communities of Seven Wild Fish Species in a Natural Lake with Limited Water Exchange during the Closed Fishing Season. Microorganisms, 12(4), 800. https://doi.org/10.3390/microorganisms12040800