
Molecular and Phylogenomic Analysis of a Vancomycin Intermediate Resistance USA300LV Strain in Chile
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain (Sampling)
2.2. Antimicrobial Susceptibility Testing
2.3. Molecular Characterization
2.3.1. Characterization of Virulence Factors
2.3.2. Multilocus Sequence Typing (MLST)
2.4. WGS and Genome Assembly
2.5. Phylogenomic Analysis
3. Results
3.1. Antimicrobial Resistance
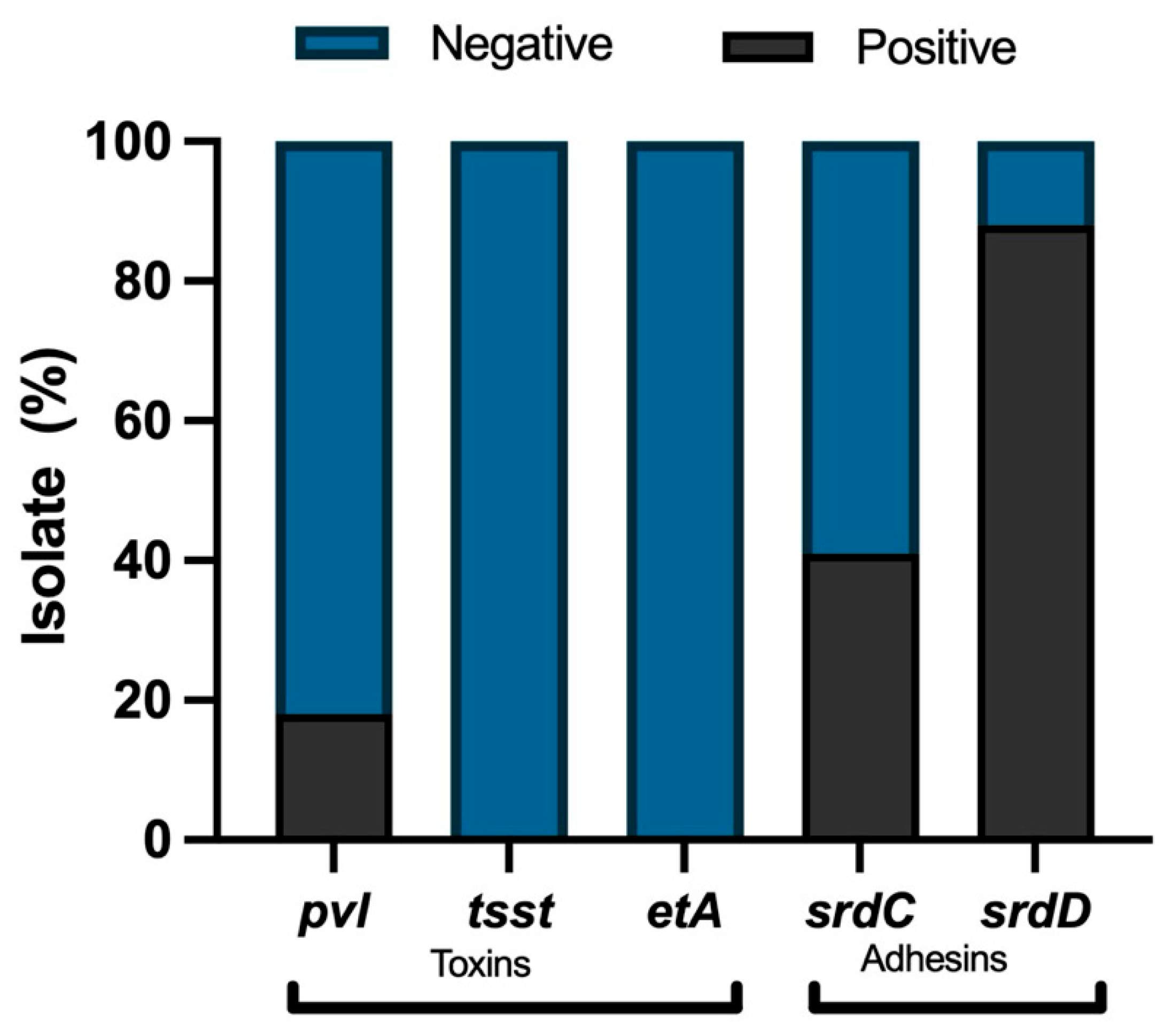
3.2. Characterization of Virulence Factors in MRSA Isolates
3.3. Multilocus Sequence Typing (MLST) of PVL-Positive Isolates
3.4. Sequencing and Genome Assembly of Isolate n°42
3.5. SCCmec Typification, Virulence Factors and Antimicrobial Resistance Profile of Isolate n°42
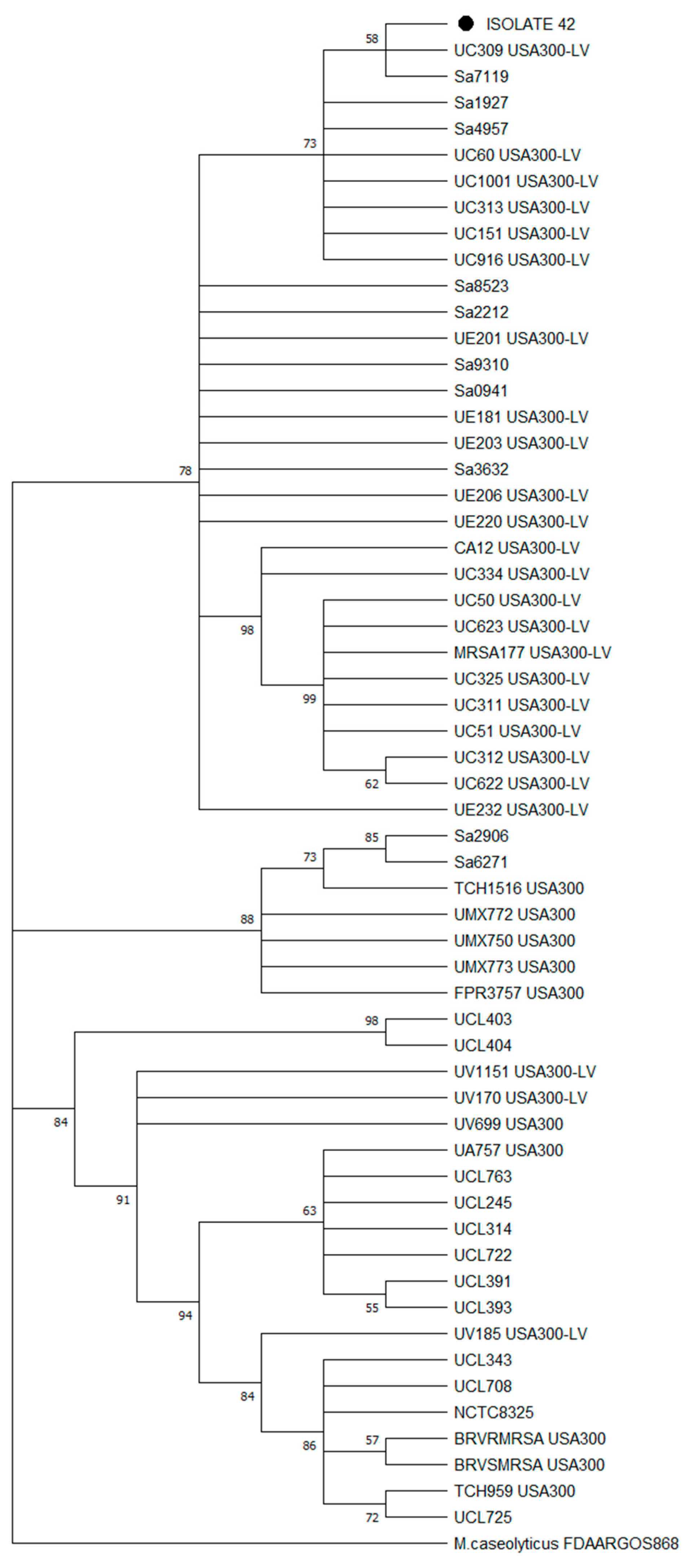
3.6. Phylogomics of Isolate n°42
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Molecular Pathogenesis of Staphylococcus aureus Infection—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/19190527/ (accessed on 19 February 2024).
- Cosgrove, S.E.; Sakoulas, G.; Perencevich, E.N.; Schwaber, M.J.; Karchmer, A.W.; Carmeli, Y. Comparison of mortality associated with methicillin-resistant and methicillin-susceptible Staphylococcus aureus bacteremia: A meta-analysis. Clin. Infect. Dis. 2003, 36, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kuwahara-Arai, K.; Katayama, Y.; Uehara, Y.; Han, X.; Kondo, Y.; Hiramatsu, K. Staphylococcal Cassette Chromosome mec (SCCmec) Analysis of MRSA. In Methicillin-Resistant Staphylococcus Aureus (MRSA) Protocols; Ji, Y., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; pp. 131–148. ISBN 978-1-62703-664-1. [Google Scholar]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31, e00020-18. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, A.; Linden, P.K.; Friedman, B. Incidence, prevalence, and management of MRSA bacteremia across patient populations—A review of recent developments in MRSA management and treatment. Crit. Care 2017, 21, 211. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, J.A.; Knight, G.M.; Budd, E.L.; McCarthy, A.J. Shuffling of mobile genetic elements (MGEs) in successful healthcare-associated MRSA (HA-MRSA). Mob. Genet. Elem. 2012, 2, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Classification of Staphylococcal Cassette Chromosome mec (SCCmec): Guidelines for Reporting Novel SCCmec Elements|Antimicrobial Agents and Chemotherapy. Available online: https://journals.asm.org/doi/10.1128/aac.00579-09 (accessed on 19 February 2024).
- Naimi, T.S.; LeDell, K.H.; Como-Sabetti, K.; Borchardt, S.M.; Boxrud, D.J.; Etienne, J.; Johnson, S.K.; Vandenesch, F.; Fridkin, S.; O’Boyle, C.; et al. Comparison of community- and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA 2003, 290, 2976–2984. [Google Scholar] [CrossRef]
- Meyer, F.; Girardot, R.; Piémont, Y.; Prévost, G.; Colin, D.A. Analysis of the specificity of Panton-Valentine leucocidin and gamma-hemolysin F component binding. Infect. Immun. 2009, 77, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Leukotoxin and Pyrogenic Toxin Superantigen Gene Backgrounds in Bloodstream and Wound Staphylococcus aureus Isolates from Eastern Region of China—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/30103694/ (accessed on 19 February 2024).
- Lo, W.-T.; Wang, C.-C. Panton-Valentine leukocidin in the pathogenesis of community-associated methicillin-resistant Staphylococcus aureus infection. Pediatr. Neonatol. 2011, 52, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Sabat, A.; Melles, D.C.; Martirosian, G.; Grundmann, H.; Van Belkum, A.; Hryniewicz, W. Distribution of the Serine-Aspartate Repeat Protein-Encoding sdr Genes among Nasal-Carriage and Invasive Staphylococcus aureus Strains. J. Clin. Microbiol. 2006, 44, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, M.; Wladyka, B.; Dubin, G. Exfoliative Toxins of Staphylococcus aureus. Toxins 2010, 2, 1148–1165. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, C.P.; Boyle-Vavra, S.; Adem, P.V.; Lee, J.C.; Husain, A.N.; Clasen, J.; Daum, R.S. Comparison of virulence in community-associated methicillin-resistant Staphylococcus aureus pulsotypes USA300 and USA400 in a rat model of pneumonia. J. Infect. Dis. 2008, 198, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.T. Community-associated methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 2005, 41 (Suppl. S4), S269–S272. [Google Scholar] [CrossRef] [PubMed]
- Kempker, R.R.; Farley, M.M.; Ladson, J.L.; Satola, S.; Ray, S.M. Association of methicillin-resistant Staphylococcus aureus (MRSA) USA300 genotype with mortality in MRSA bacteremia. J. Infect. 2010, 61, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Como-Sabetti, K.; Harriman, K.H.; Buck, J.M.; Glennen, A.; Boxrud, D.J.; Lynfield, R. Community-associated methicillin-resistant Staphylococcus aureus: Trends in case and isolate characteristics from six years of prospective surveillance. Public. Health Rep. 2009, 124, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Planet, P.J.; Diaz, L.; Kolokotronis, S.-O.; Narechania, A.; Reyes, J.; Xing, G.; Rincon, S.; Smith, H.; Panesso, D.; Ryan, C.; et al. Parallel Epidemics of Community-Associated Methicillin-Resistant Staphylococcus aureus USA300 Infection in North and South America. J. Infect. Dis. 2015, 212, 1874–1882. [Google Scholar] [CrossRef]
- Arias, C.A.; Rincon, S.; Chowdhury, S.; Martínez, E.; Coronell, W.; Reyes, J.; Nallapareddy, S.R.; Murray, B.E. MRSA USA300 clone and VREF—A U.S.-Colombian connection? N. Engl. J. Med. 2008, 359, 2177–2179. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.A.; Reyes, J.; Carvajal, L.P.; Rincon, S.; Diaz, L.; Panesso, D.; Ibarra, G.; Rios, R.; Munita, J.M.; Salles, M.J.; et al. A Prospective Cohort Multicenter Study of Molecular Epidemiology and Phylogenomics of Staphylococcus aureus Bacteremia in Nine Latin American Countries. Antimicrob. Agents Chemother. 2017, 61, e00816-17. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.R.W.; Planet, P.J.; Spencer-Sandino, M.; Rivas, L.; Díaz, L.; Moustafa, A.M.; Quesille-Villalobos, A.; Riquelme-Neira, R.; Alcalde-Rico, M.; Hanson, B.; et al. Dynamics of the MRSA Population in a Chilean Hospital: A Phylogenomic Analysis (2000–2016). Microbiol. Spectr. 2023, 11, e0535122. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.M.; Moustafa, A.M.; O’Brien, K.; Martin, M.A.; Read, T.D.; Kreiswirth, B.N.; Planet, P.J. Pre-epidemic evolution of the MRSA USA300 clade and a molecular key for classification. Front. Cell. Infect. Microbiol. 2023, 13, 1081070. [Google Scholar] [CrossRef] [PubMed]
- Medina, G.; Egea, A.L.; Otth, C.; Otth, L.; Fernández, H.; Bocco, J.L.; Wilson, M.; Sola, C. Molecular epidemiology of hospital-onset methicillin-resistant Staphylococcus aureus infections in Southern Chile. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- CLSI-2020.pdf. Available online: https://www.nih.org.pk/wp-content/uploads/2021/02/CLSI-2020.pdf (accessed on 6 May 2024).
- Bhattacharya, S. Evaluation of Multidrug Resistant Staphylococcus aureus and their Association with Biofilm Production in a Tertiary Care Hospital, Tripura, Northeast India. JCDR 2015, 9, DC01. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.L.; Goldman, J.; Sherman, A.K.; Jeremiah Bell, J.; Selveraju, S.; Newland, J.G.; Jarka, D.E.; Chastain, K.; Selvarangan, R. Clinical variables and Staphylococcus aureus virulence factors associated with venous thromboembolism in children. Thromb. Res. 2016, 138, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Enright, M.C.; Day, N.P.J.; Davies, C.E.; Peacock, S.J.; Spratt, B.G. Multilocus Sequence Typing for Characterization of Methicillin-Resistant and Methicillin-Susceptible Clones of Staphylococcus aureus. J. Clin. Microbiol. 2000, 38, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, M.; Wildenhain, J.; Rappsilber, J.; Tyers, M. BoxPlotR: A web tool for generation of box plots. Nat. Methods 2014, 11, 121–122. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucl. Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Cumsille, A.; Durán, R.E.; Rodríguez-Delherbe, A.; Saona-Urmeneta, V.; Cámara, B.; Seeger, M.; Araya, M.; Jara, N.; Buil-Aranda, C. GenoVi, an open-source automated circular genome visualizer for bacteria and archaea. PLoS Comput. Biol. 2023, 19, e1010998. [Google Scholar] [CrossRef]
- Kaya, H.; Hasman, H.; Larsen, J.; Stegger, M.; Johannesen, T.B.; Allesøe, R.L.; Lemvigh, C.K.; Aarestrup, F.M.; Lund, O.; Larsen, A.R. SCC mec Finder, a Web-Based Tool for Typing of Staphylococcal Cassette Chromosome mec in Staphylococcus aureus Using Whole-Genome Sequence Data. mSphere 2018, 3, e00612-17. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. The UGENE team Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-Time Whole-Genome Sequencing for Routine Typing, Surveillance, and Outbreak Detection of Verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef]
- Malberg Tetzschner, A.M.; Johnson, J.R.; Johnston, B.D.; Lund, O.; Scheutz, F. In Silico Genotyping of Escherichia coli Isolates for Extraintestinal Virulence Genes by Use of Whole-Genome Sequencing Data. J. Clin. Microbiol. 2020, 58, e01269-20. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.D.; Negi, Y.K.; Gaur, A.; Khanna, D. Detection of virulence genes in Staphylococcus aureus isolated from paper currency. Int. J. Infect. Dis. 2009, 13, e450–e455. [Google Scholar] [CrossRef] [PubMed]
- Seas, C.; Garcia, C.; Salles, M.J.; Labarca, J.; Luna, C.; Alvarez-Moreno, C.; Mejía-Villatoro, C.; Zurita, J.; Guzmán-Blanco, M.; Rodríguez-Noriega, E.; et al. Staphylococcus aureus bloodstream infections in Latin America: Results of a multinational prospective cohort study. J. Antimicrob. Chemother. 2018, 73, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J. Statistical Brief #212Hospital-, Health Care-, and Community-Acquired MRSA: Estimates from California Hospitals. 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK396238/ (accessed on 7 March 2023).
- Saeed, K.; Ahmad, N.; Dryden, M.; Cortes, N.; Marsh, P.; Sitjar, A.; Wyllie, S.; Bourne, S.; Hemming, J.; Jeppesen, C.; et al. Oxacillin-susceptible methicillin-resistant Staphylococcus aureus (OS-MRSA), a hidden resistant mechanism among clinically significant isolates in the Wessex region/UK. Infection 2014, 42, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Cuirolo, A.; Canigia, L.F.; Gardella, N.; Fernández, S.; Gutkind, G.; Rosato, A.; Mollerach, M. Oxacillin- and cefoxitin-susceptible meticillin-resistant Staphylococcus aureus (MRSA). Int. J. Antimicrob. Agents 2011, 37, 178–179. [Google Scholar] [CrossRef]
- Li, X.; Huang, T.; Xu, K.; Li, C.; Li, Y. Molecular characteristics and virulence gene profiles of Staphylococcus aureus isolates in Hainan, China. BMC Infect. Dis. 2019, 19, 873. [Google Scholar] [CrossRef] [PubMed]
- Shariati, A.; Dadashi, M.; Moghadam, M.T.; van Belkum, A.; Yaslianifard, S.; Darban-Sarokhalil, D. Global prevalence and distribution of vancomycin resistant, vancomycin intermediate and heterogeneously vancomycin intermediate Staphylococcus aureus clinical isolates: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 12689. [Google Scholar] [CrossRef]
- ISP. Vigilancia de Resistencia a Vancomicina en Bacterias que Pueden Producir Infecciones Asociadas a la Atención en Salud (IAAS); ISP: Santiago, Chile, 2019; Volume 9. [Google Scholar]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Preeja, P.P.; Kumar, S.H.; Shetty, V. Prevalence and Characterization of Methicillin-Resistant Staphylococcus aureus from Community- and Hospital-Associated Infections: A Tertiary Care Center Study. Antibiotics 2021, 10, 197. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.P.; Park, S.-J.; Kim, H.S.; Kim, E.S.; Kim, M.-N.; Park, K.-H.; Kim, S.-H.; Lee, S.-O.; Choi, S.-H.; Jeong, J.-Y.; et al. Persistent Staphylococcus aureus Bacteremia: A Prospective Analysis of Risk Factors, Outcomes, and Microbiologic and Genotypic Characteristics of Isolates. Medicine 2013, 92, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Paniagua-Contreras, G.; Sáinz-Espuñes, T.; Monroy-Pérez, E.; Raymundo Rodríguez-Moctezuma, J.; Arenas-Aranda, D.; Negrete-Abascal, E.; Vaca, S. Virulence Markers in Staphylococcus aureus Strains Isolated from Hemodialysis Catheters of Mexican Patients. AiM 2012, 2, 476–487. [Google Scholar] [CrossRef]
- Perbet, S.; Soummer, A.; Vinsonneau, C.; Vandebrouck, A.; Rackelboom, T.; Etienne, J.; Cariou, A.; Chiche, J.-D.; Mira, J.-P.; Charpentier, J. Multifocal community-acquired necrotizing fasciitis caused by a Panton-Valentine leukocidin-producing methicillin-sensitive Staphylococcus aureus. Infection 2010, 38, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.R.W.; Diaz, L.; Rojas, M.; Rios, R.; Hanson, B.; Rivas, L.M.; Spencer, M.; Moustafa, A.M.; Araos Bralic, R.; Peters, A.; et al. Phylogenomic Epidemiology of Methicillin-Resistant Staphylococcus aureus (MRSA) Chilean-Cordobes Clone in Latin America. Open Forum Infect. Dis. 2019, 6, S263–S264. [Google Scholar] [CrossRef]
- Noriega, L.M.; González, P.; Hormazábal, J.C.; Pinto, C.; Canals, M.; Munita, J.M.; Thompson, L.; Marcotti, A.; Pérez, J.; Ibáñez, D.; et al. Staphylococcus aureus comunitario resistente a cloxacilina: Comunicación de los primeros cinco casos descritos en Chile. Rev. Méd. Chile 2008, 136, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.L.; O’Hara, F.P.; Close, N.M.; Mera, R.M.; Miller, L.A.; Suaya, J.A.; Amrine-Madsen, H. Prevalence and Sequence Variation of Panton-Valentine Leukocidin in Methicillin-Resistant and Methicillin-Susceptible Staphylococcus aureus Strains in the United States. J. Clin. Microbiol. 2012, 50, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Prévost, G.; Cribier, B.; Couppié, P.; Petiau, P.; Supersac, G.; Finck-Barbançon, V.; Monteil, H.; Piemont, Y. Panton-Valentine leucocidin and gamma-hemolysin from Staphylococcus aureus ATCC 49775 are encoded by distinct genetic loci and have different biological activities. Infect Immun 1995, 63, 4121–4129. [Google Scholar] [CrossRef]
- Albrecht, N.; Jatzwauk, L.; Slickers, P.; Ehricht, R.; Monecke, S. Clonal Replacement of Epidemic Methicillin-Resistant Staphylococcus aureus Strains in a German University Hospital over a Period of Eleven Years. PLoS ONE 2011, 6, e28189. [Google Scholar] [CrossRef] [PubMed]
- Sassi, M.; Felden, B.; Revest, M.; Tattevin, P.; Augagneur, Y.; Donnio, P.-Y. An outbreak in intravenous drug users due to USA300 Latin-American variant community-acquired methicillin-resistant Staphylococcus aureus in France as early as 2007. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2495–2501. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, A.M.; Vélez, L.A.; Robledo, J.; Jiménez, J.N. Cambios a lo largo del tiempo en la distribución de los complejos de clones dominantes de Staphylococcus aureus resistente a la meticilina en Medellín, Colombia. BioMedica 2013, 34, 34. [Google Scholar] [CrossRef]
- Aguayo-Reyes, A.; Quezada-Aguiluz, M.; Mella, S.; Riedel, G.; Opazo-Capurro, A.; Bello-Toledo, H.; Domínguez, M.; González-Rocha, G. Bases moleculares de la resistencia a meticilina en Staphylococcus aureus. Rev. Chil. Infectol. 2018, 35, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Shokrollahi, P.; Hasani, A.; Aghazadeh, M.; Memar, M.Y.; Hasani, A.; Zaree, M.; Rezaee, M.A.; Sadeghi, J. Contribution of Arginine Catabolic Mobile Element and Copper and Mercury Resistance Element in Methicillin-Resistant Staphylococcus aureus: A Vantage Point. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 9916255. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-C.; Jeon, B. Novel adjuvant strategy to potentiate bacitracin against MDR MRSA. J. Antimicrob. Chemother. 2016, 71, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Yamada, K.; Nagao, M.; Aoki, E.; Matsumoto, M.; Hirayama, T.; Yamamoto, H.; Hiramatsu, R.; Ichiyama, S.; Iinuma, Y. Antimicrobial Ointments and Methicillin-Resistant Staphylococcus aureus USA300. Emerg. Infect. Dis. 2011, 17, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, J.; Zhang, Y.; Wang, D.; Liu, R.; Liu, G.; Yao, H.; Pan, Z. Bacitracin resistance and enhanced virulence of Streptococcus suis via a novel efflux pump. BMC Vet. Res. 2019, 15, 377. [Google Scholar] [CrossRef] [PubMed]
- Otarigho, B.; Falade, M.O. Analysis of antibiotics resistant genes in different strains of Staphylococcus aureus. Bioinformation 2018, 14, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Velhner, M.; Milanov, D. Resistance to tetracycline in Escherichia coli and Staphylococcus aureus: Brief overview on mechanisms of resistance and epidemiology. AVM 2016, 8, 27–36. [Google Scholar] [CrossRef]
- Wang, W.; Sun, B. VraCP regulates cell wall metabolism and antibiotic resistance in vancomycin-intermediate Staphylococcus aureus strain Mu50. J. Antimicrob. Chemother. 2021, 76, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, W.A.; Malachowa, N.; DeLeo, F.R. Vancomycin Resistance in Staphylococcus aureus. Yale J. Biol. Med. 2017, 90, 269–281. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic | MRSA n = 51 (%) | ||
|---|---|---|---|
| R | I | S | |
| Oxacillin | 48 (94.1) | - | 3 (5.9) |
| Vancomycin | - | 1(2.0) | 50 (98.0) |
| Ciprofloxacin | 43 (84.3) | 2 (3.9) | 6 (11.8) |
| Chloramphenicol | 2 (3.9) | 1 (2.0) | 48 (94.1) |
| Gentamicin | 25 (49.0) | - | 26 (51.0) |
| Levofloxacin | 44 (86.3) | 1 (2.0) | 6 (11.8) |
| Norfloxacin | 45 (88.2) | 1 (2.0) | 5 (9.8) |
| Rifampin | 6 (11.8) | - | 45 (88.2) |
| Tetracycline | - | - | 51 (100) |
| Isolate No. | ST (CC) | Antibiotic Resistance Phenotype | Virulence Factors |
|---|---|---|---|
| 11 | 5 (5) | OXA, GEN, CIP, LEV, NOR | sdrC, sdrD, pvl |
| 12 | 923 (8) | OXA | sdrC, sdrD, pvl |
| 13 | 5 (5) | OXA, GEN, CIP, NOR | sdrC, sdrD, pvl |
| 14 | 72 (8) | OXA, CIP, LEV, NOR, RIF | sdrD, pvl |
| 38 | 8 (8) | OXA, CHLO, CIP, LEV, NOR | sdrD, pvl |
| 42 | 8 (8) | OXA, Vancomycin intermediate (VISA) | sdrC, sdrD, pvl |
| 43 | 8 (8) | OXA | sdrD, pvl |
| 44 | 5 (5) | OXA, GEN, CIP, LEV, NOR, RIF | sdrD, pvl |
| 45 | 5 (5) | OXA, GEN, CIP, LEV, NOR, RIF | sdrD, pvl |
| ID Sequence | Phred 30 Average Quality | Number of Reads (Millions) | Reads Length (bp) |
|---|---|---|---|
| 42_S34_R1_001 | 33.5 | 8.1 | 149 |
| 42_S34_R2_001 | 33.2 | 8.1 | 148 |
| 42_S34_trimmed_1P | 34.7 | 6.3 | 129 |
| 42_S34_trimmed_2P | 34.6 | 6.3 | 123 |
| Genome Feature | Result |
|---|---|
| Average coverage | 290X |
| k-mer size used by Velvet assembler | 111 bp |
| N50 | 453,788 bp |
| L50 | 3 |
| Number of contigs | 49 |
| Genome size | 2,800,637 bp |
| Number of protein-coding genes | 2574 |
| GC content | 32% |
| Complete and single copy BUSCOs—Domain | 100% |
| Complete and single copy BUSCOs—Phylum | 99.5% |
| Complete and single copy BUSCOs—Class | 99.7% |
| Complete and single copy BUSCOs—Order | 100 |
| 23S rRNA genes | 2 |
| 16S rRNA genes | 3 |
| 5S rRNA genes | 7 |
| tRNA genes | 62 |
| Gene | Encoded Protein |
|---|---|
| hlgA | Gamma-hemolysin chain II precursor |
| hlgB | Precursor of component B gamma-hemolysin |
| hlgC | Gamma-hemolysin component C |
| lukD | Leukocidin D component |
| lukE | Leukocidin E component |
| lukF-PV | Panton Valentine Leukocidin F Component |
| lukS-PV | Panton Valentine Leukocidin S Component |
| sek | Enterotoxin K |
| seq | Enterotoxin Q |
| aur | Aureolysin |
| splA | Serine protease splA |
| splB | Serine protease splB |
| splE | Serine protease splE |
| spa | Staphylococcal protein A |
| sdrC | Serine-aspartate repeat proteins |
| sdrD | Serine-aspartate repeat proteins |
| sdrE | Serine-aspartate repeat proteins |
| sasA | Staphylococcus aureus surface protein A |
| sasC | Staphylococcus aureus surface protein C |
| sasD | Staphylococcus aureus surface protein D |
| sasF | Staphylococcus aureus surface protein F |
| sasG | Staphylococcus aureus surface protein G |
| sasH | Staphylococcus aureus surface protein H |
| fnbA | Fibronectin binding protein A |
| fnbB | Fibronectin binding protein B |
| efb | Extracellular fibrinogen-binding protein |
| clfA | Clumping factor A |
| Antimicrobial | Gene |
|---|---|
| Bacitracin | bceA |
| bceB | |
| bceR | |
| bceS | |
| Quinolones | gyrA |
| gyrB | |
| norB | |
| Tetracycline | tetR |
| tetA | |
| Vancomycin | vraR |
| vraS | |
| Fosfomycin | fosB |
| Bicozamycin | bcr |
| β-lactam | mecA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Núñez, D.; Jiménez, P.; Cortez-San Martín, M.; Cortés, C.; Cárdenas, M.; Michelson, S.; Garay, T.; Vecchiola, M.; Céspedes, A.; Maldonado, J.E.; et al. Molecular and Phylogenomic Analysis of a Vancomycin Intermediate Resistance USA300LV Strain in Chile. Microorganisms 2024, 12, 1284. https://doi.org/10.3390/microorganisms12071284
Núñez D, Jiménez P, Cortez-San Martín M, Cortés C, Cárdenas M, Michelson S, Garay T, Vecchiola M, Céspedes A, Maldonado JE, et al. Molecular and Phylogenomic Analysis of a Vancomycin Intermediate Resistance USA300LV Strain in Chile. Microorganisms. 2024; 12(7):1284. https://doi.org/10.3390/microorganisms12071284
Chicago/Turabian StyleNúñez, Daniela, Pablo Jiménez, Marcelo Cortez-San Martín, Carolina Cortés, Matías Cárdenas, Sofia Michelson, Tamara Garay, Maggie Vecchiola, Alejandra Céspedes, Jonathan E. Maldonado, and et al. 2024. "Molecular and Phylogenomic Analysis of a Vancomycin Intermediate Resistance USA300LV Strain in Chile" Microorganisms 12, no. 7: 1284. https://doi.org/10.3390/microorganisms12071284