
Population Genomics analysis of Leptosphaeria biglobosa Associated with Brassica napus in China Reveals That Geographical Distribution Influences Its Genetic Polymorphism

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strains and DNA Extraction
2.2. Genome Sequencing
2.3. Genome Assembly and Annotation
2.4. SNP Calling and Filtering
2.5. Population Structure and Principal Component Analysis
2.6. Phylogenetic Tree
2.7. Nucleotide Diversity and Fst
2.8. Linkage Disequilibrium
3. Results
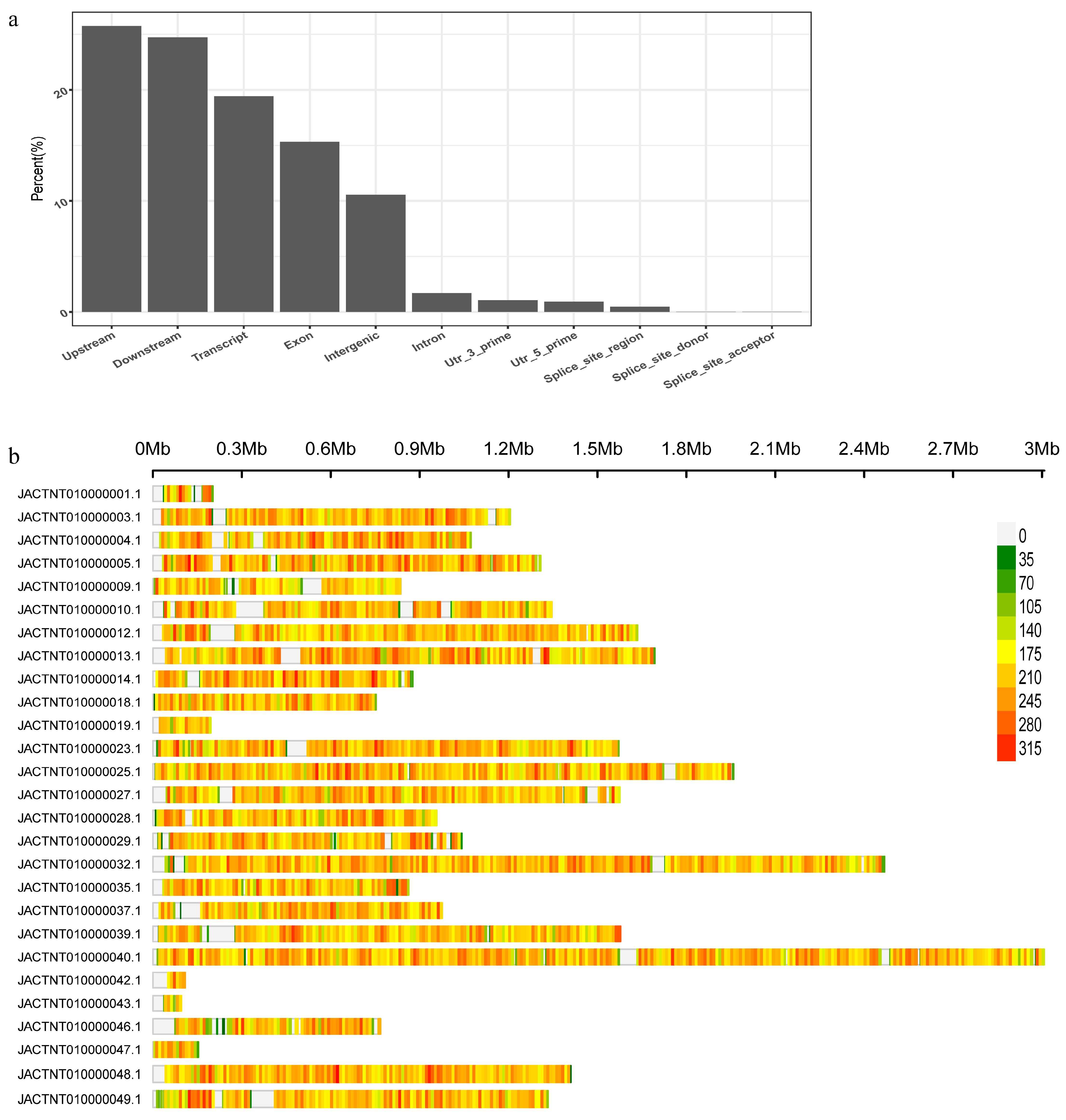
3.1. Genome Assembly and Annotatiopn
3.2. Sequencing, SNP Calling, and Read Mapping
3.3. Population Structure and Principal Component Analysis
3.4. Phylogenetic Tree
3.5. Genome Diversity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cai, X.; Yang, L.; Zhang, J.; Li, G.Q. First Report of Leptosphaeria biglobosa Causing Black Leg on Brassica campestris ssp. chinensis var. purpurea in Central China. Plant Dis. 2014, 98, 1156. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Li, G.; Yang, L. First report of Leptosphaeria biglobosa ‘canadensis’ causing blackleg on oilseed rape (Brassica napus) in China. Plant Dis. 2021, 105, 3760. [Google Scholar] [CrossRef] [PubMed]
- Magyar, D.; Barasits, T.; Fischl, G.; Fernando, W.G.D. First Report of the Natural Occurrence of the Teleomorph of Leptosphaeria maculans on Oilseed Rape and Airborne Dispersal of Ascospores in Hungary. J. Phytopathol. 2010, 154, 428–431. [Google Scholar] [CrossRef]
- Akhatar, J.; Singh, M.P.; Sharma, A.; Kaur, H.; Kaur, N.; Sharma, S.; Bharti, B.; Sardana, V.; Banga, S.S. Association mapping of seed quality traits under varying conditions of nitrogen application in Brassica juncea L. Czern & Coss. Front. Genet. 2020, 11, 744. [Google Scholar] [CrossRef]
- Van de Wouw, A.P.; Marcroft, S.J.; Sprague, S.J.; Scanlan, J.L.; Vesk, P.A.; Idnurm, A. Epidemiology and management of blackleg of canola in response to changing farming practices in Australia. Australas Plant Path 2021, 50, 137–149. [Google Scholar] [CrossRef]
- Huang, Y.J.; Sidique, S.N.M.; Dewage, C.S.K.; Gajula, L.H.; Mitrousia, G.K.; Qi, A.; West, J.S.; Fitt, B.D. Effective control of Leptosphaeria maculans increases importance of L. biglobosa as a cause of phoma stem canker epidemics on oilseed rape. Pest Manag. Sci. 2024, 80, 2405–2415. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Strelkov, S.E.; Hwang, S.F. Blackleg Yield Losses and Interactions with Verticillium Stripe in Canola (Brassica napus) in Canada. Plants 2023, 12, 434. [Google Scholar] [CrossRef] [PubMed]
- Balesdent, M.H.; Laval, V.; Noah, J.M.; Bagot, P.; Mousseau, A.; Rouxel, T. Large-scale population survey of Leptosphaeria maculans in France highlights both on-going breakdowns and potentially effective resistance genes in oilseed rape. Pest Manag. Sci. 2024, 80, 2426–2434. [Google Scholar] [CrossRef] [PubMed]
- Barnes, A.P.; Wreford, A.; Butterworth, M.H.; Semenov, M.A.; Fitt, B.D.L. Adaptation to increasing severity of phoma stem canker on winter oilseed rape in the UK under climate change. J. Agric. Sci. 2010, 148, 683–694. [Google Scholar] [CrossRef]
- Wang, Y.X.; Strelkov, S.E.; Hwang, S.F. Yield losses in canola in response to blackleg disease. Can. J. Plant Sci. 2020, 100, 488–494. [Google Scholar] [CrossRef]
- Ghanbarnia, K.; Fudal, I.; Larkan, N.J.; Links, M.G.; Balesdent, M.H.; Profotova, B.; Fernando, W.G.; Rouxel, T.; Borhan, M.H. Rapid identification of the Leptosphaeria maculans avirulence gene AvrLm2 using an intraspecific comparative genomics approach. Mol. Plant Pathol. 2015, 16, 699–709. [Google Scholar] [CrossRef]
- Lowe, R.G.; Cassin, A.; Grandaubert, J.; Clark, B.L.; Van de Wouw, A.P.; Rouxel, T.; Howlett, B.J. Genomes and transcriptomes of partners in plant-fungal-interactions between canola (Brassica napus) and two Leptosphaeria species. PLoS ONE 2014, 9, e103098. [Google Scholar] [CrossRef]
- West, J.S.; Balesdent, M.H.; Rouxel, T.; Narcy, J.P.; Huang, Y.J.; Roux, J.; Steed, J.M.; Fitt, B.D.L.; Schmit, J. Colonization of winter oilseed rape tissues by A/Tox+ and B/Tox0Leptosphaeria maculans (phoma stem canker) in France and England. Plant Pathol. 2002, 51, 311–321. [Google Scholar] [CrossRef]
- Fitt, B.D.L.; Brun, H.; Barbetti, M.J.; Rimmer, S.R. World-Wide Importance of Phoma Stem Canker (Leptosphaeria maculans and L. biglobosa) on Oilseed Rape (Brassica napus). Eur. J. Plant Pathol. 2006, 114, 3–15. [Google Scholar] [CrossRef]
- van de Wouw, A.P.; Scanlan, J.L.; Al-Mamun, H.A.; Balesdent, M.H.; Bousset, L.; Burketová, L.; Mendoza, L.D.; Fernando, W.G.D.; Franke, C.; Howlett, B.J.; et al. A new set of international Leptosphaeria maculans isolates as a resource for elucidation of the basis and evolution of blackleg disease on Brassica napus. Plant Pathol. 2024, 73, 170–185. [Google Scholar] [CrossRef]
- Deng, Y.; Li, J.C.; Lyv, X.; Xu, J.W.; Wu, M.D.; Zhang, J.; Yang, L.; Li, G.Q. Large-Scale Surveys of Blackleg of Oilseed Rape (Leptosphaeria biglobosa) Revealed New Insights into Epidemics of This Disease in China. Plant Dis. 2023, 107, 1408–1417. [Google Scholar] [CrossRef]
- Long, Y.; Shang, M.; Deng, Y.; Yu, C.; Wu, M.; Li, G. First Report of Leptosphaeria biglobosa ‘brassicae’ Causing Blackleg on Brassica juncea var. multisecta in China. Plant Dis. 2021, 105, 3749. [Google Scholar] [CrossRef]
- West, J.S.; Evans, N.; Liu, S.; Hu, B.; Peng, L. Leptosphaeria maculans causing stem canker of oilseed rape in China. Plant Pathol. 2000, 49, 800. [Google Scholar] [CrossRef]
- Lei, R.; Kong, J.; Qiu, Y.; Chen, N.; Zhu, S.; Wang, X.; Wu, P. Rapid detection of the pathogenic fungi causing blackleg of Brassica napus using a portable real-time fluorescence detector. Food Chem. 2019, 288, 57–67. [Google Scholar] [CrossRef]
- Dutreux, F.; Da Silva, C.; d’Agata, L.; Couloux, A.; Gay, E.J.; Istace, B.; Lapalu, N.; Lemainque, A.; Linglin, J.; Noel, B.; et al. De novo assembly and annotation of three Leptosphaeria genomes using Oxford Nanopore MinION sequencing. Sci. Data 2018, 5, 180235. [Google Scholar] [CrossRef]
- Liu, Z.; Latunde-Dada, A.O.; Hall, A.M.; Fitt, B.D.L. Phoma stem canker disease on oilseed rape (Brassica napus) in China is caused by Leptosphaeria biglobosa ‘brassicae’. Eur. J. Plant Pathol. 2014, 140, 841–857. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. A J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef]
- Massouras, A.; Hens, K.; Gubelmann, C.; Uplekar, S.; Decouttere, F.; Rougemont, J.; Cole, S.T.; Deplancke, B. Primer-initiated sequence synthesis to detect and assemble structural variants. Nat. Methods 2010, 7, 485–486. [Google Scholar] [CrossRef]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.11–14.10.14. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of Apoplastic and Cytoplasmic Effectors in Fungi and Oomycetes. Mol. Plant-Microbe Interact. MPMI 2022, 35, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017, bioRxiv:201178. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2018, 35, 1786–1788. [Google Scholar] [CrossRef]
- Rouxel, T.; Balesdent, M.H. The stem canker (blackleg) fungus, Leptosphaeria maculans, enters the genomic era. Mol. Plant Pathol. 2005, 6, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Zhang, J.; Yang, L.; Li, G.Q.; Wu, M.D. Genetic Diversity and Population Structure of Leptosphaeria biglobosa from the Winter Oilseed Rape Region in China. J. Fungi 2023, 9, 1092. [Google Scholar] [CrossRef] [PubMed]
- Delihas, N. Impact of small repeat sequences on bacterial genome evolution. Genome Biol. Evol. 2011, 3, 959–973. [Google Scholar] [CrossRef]
- Liao, X.; Zhu, W.; Zhou, J.; Li, H.; Xu, X.; Zhang, B.; Gao, X. Repetitive DNA sequence detection and its role in the human genome. Commun. Biol. 2023, 6, 954. [Google Scholar] [CrossRef]
- Welgemoed, T.; Duong, T.A.; Barnes, I.; Stukenbrock, E.H.; Berger, D.K. Population genomic analyses suggest recent dispersal events of the pathogen Cercospora zeina into East and Southern African maize cropping systems. G3 Genes. 2023, 13, jkad214. [Google Scholar] [CrossRef]
- Chen, Q.L.; Peng, G.; Kutcher, R.; Yu, F.Q. Genetic diversity and population structure of Leptosphaeria maculans isolates in Western Canada. J. Genet. Genom. 2021, 48, 994–1006. [Google Scholar] [CrossRef]
- Hao, L.; Song, P.; Huangfu, H.; Li, Z. Genetic diversity and differentiation of Leptosphaeria biglobosa on oilseed rape in China. Phytoparasitica 2014, 43, 253–263. [Google Scholar] [CrossRef]
- Hao, L.; Song, P.; Huangfu, H.; Li, Z. Genetic diversity of phoma stem canker pathogen Leptosphaeria biglobosa by ISSR. Chin. J. Oil Crop. Sci. 2014, 43, 253–263. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; Guo, Z.; Bao, S.; Xu, J.; Li, K.; Rong, S.; Li, Q.; Xu, A.; Zhandui, D.; Huang, Z.; et al. Population Genomics analysis of Leptosphaeria biglobosa Associated with Brassica napus in China Reveals That Geographical Distribution Influences Its Genetic Polymorphism. Microorganisms 2024, 12, 1347. https://doi.org/10.3390/microorganisms12071347
Shi Y, Guo Z, Bao S, Xu J, Li K, Rong S, Li Q, Xu A, Zhandui D, Huang Z, et al. Population Genomics analysis of Leptosphaeria biglobosa Associated with Brassica napus in China Reveals That Geographical Distribution Influences Its Genetic Polymorphism. Microorganisms. 2024; 12(7):1347. https://doi.org/10.3390/microorganisms12071347
Chicago/Turabian StyleShi, Yiji, Zhiting Guo, Shunjun Bao, Jiali Xu, Keqi Li, Songbai Rong, Qiangsheng Li, Aixia Xu, Duojie Zhandui, Zhen Huang, and et al. 2024. "Population Genomics analysis of Leptosphaeria biglobosa Associated with Brassica napus in China Reveals That Geographical Distribution Influences Its Genetic Polymorphism" Microorganisms 12, no. 7: 1347. https://doi.org/10.3390/microorganisms12071347