
Comparative Metagenomic Analysis Reveals Rhizosphere Microbiome Assembly and Functional Adaptation Changes Caused by Clubroot Disease in Chinese Cabbage

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Field Experiment and Sample Collection
2.2. Physiochemical Analysis
2.3. DNA Extraction and Sequencing
2.4. Metagenomic Data Analysis
2.5. Taxonomic Predictions and Functional Annotations
2.6. Statistical Analysis
3. Results
3.1. General Characteristics of Metagenome Illumina PE150 Sequencing Results
3.2. Gene Prediction and Abundance Analysis
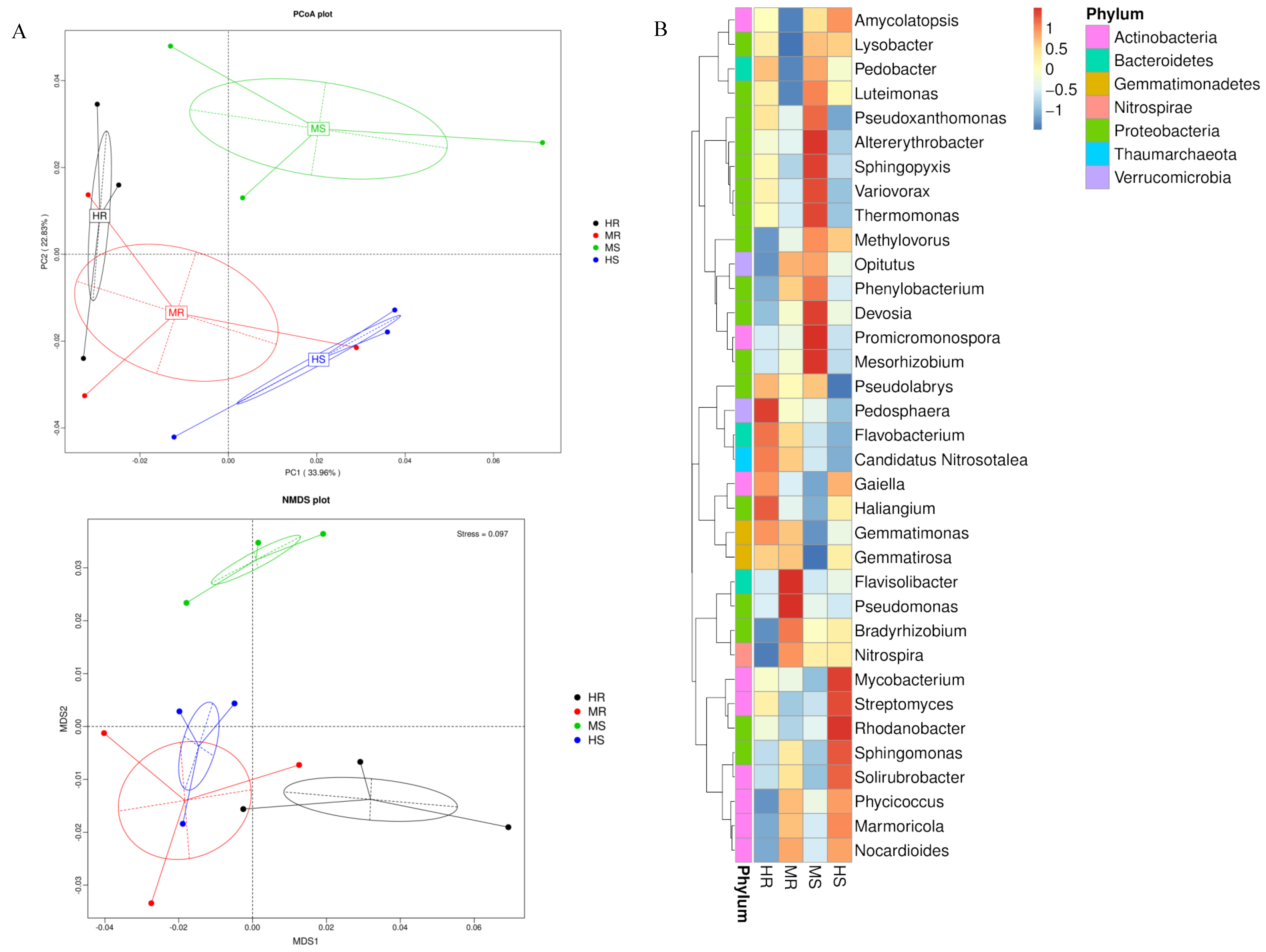
3.3. Analysis of the Microbial Community Composition
3.4. Multiple Soil Environmental Factors Significantly Correlated with Clubroot Disease
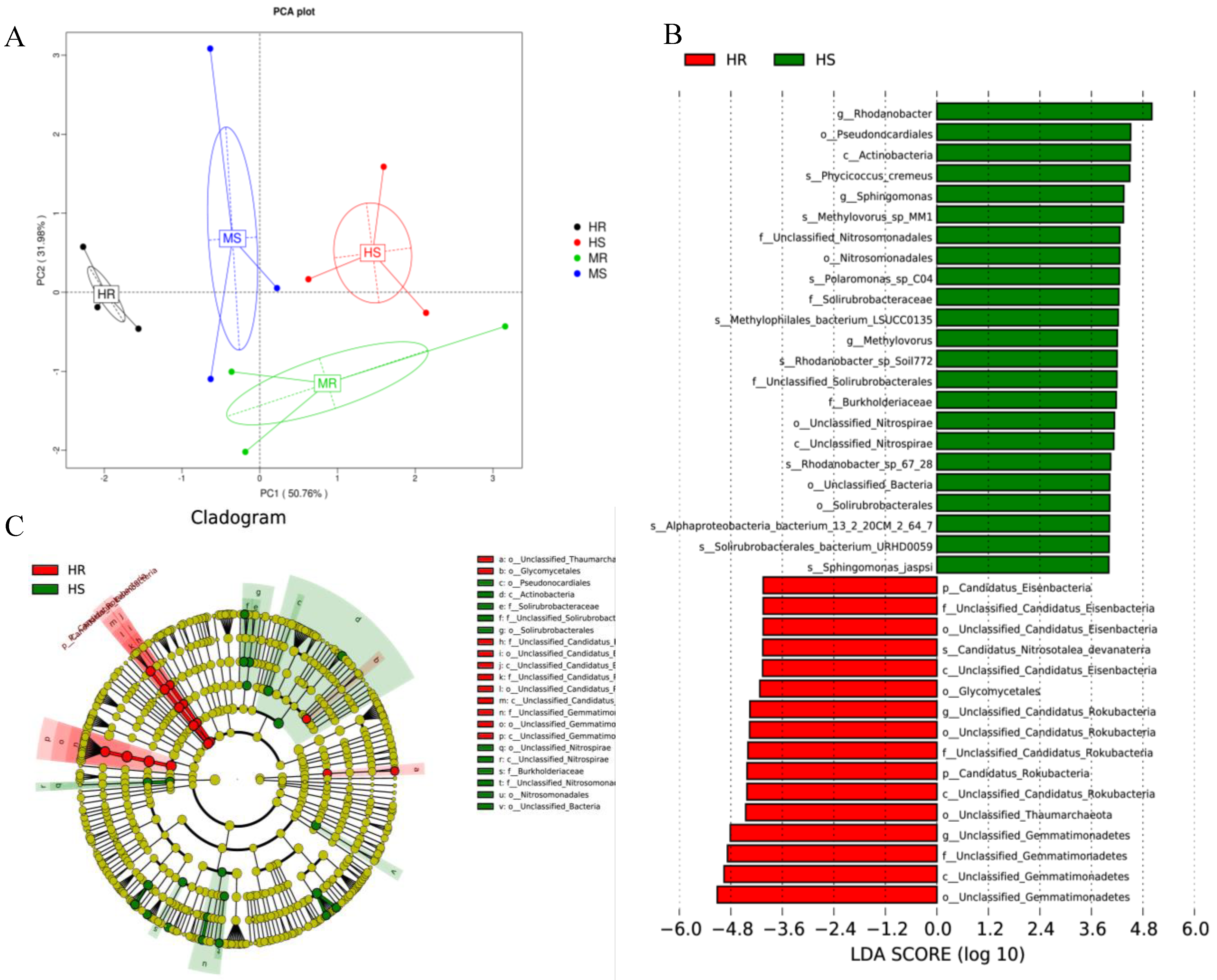
3.5. Changes in Core and Specific Microbes after Pathogen Infection
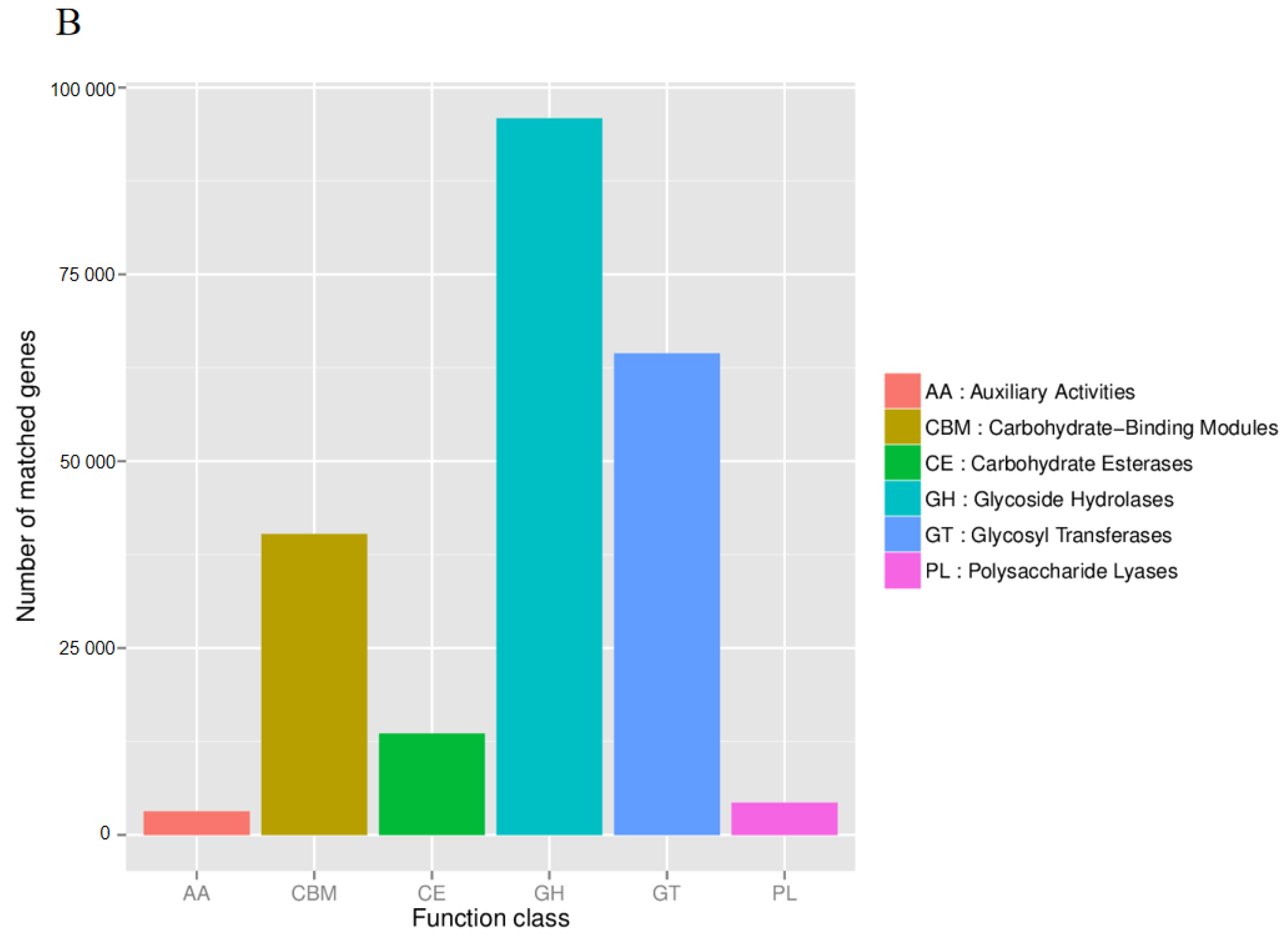
3.6. Functional Composition of Rhizosphere Microbiota
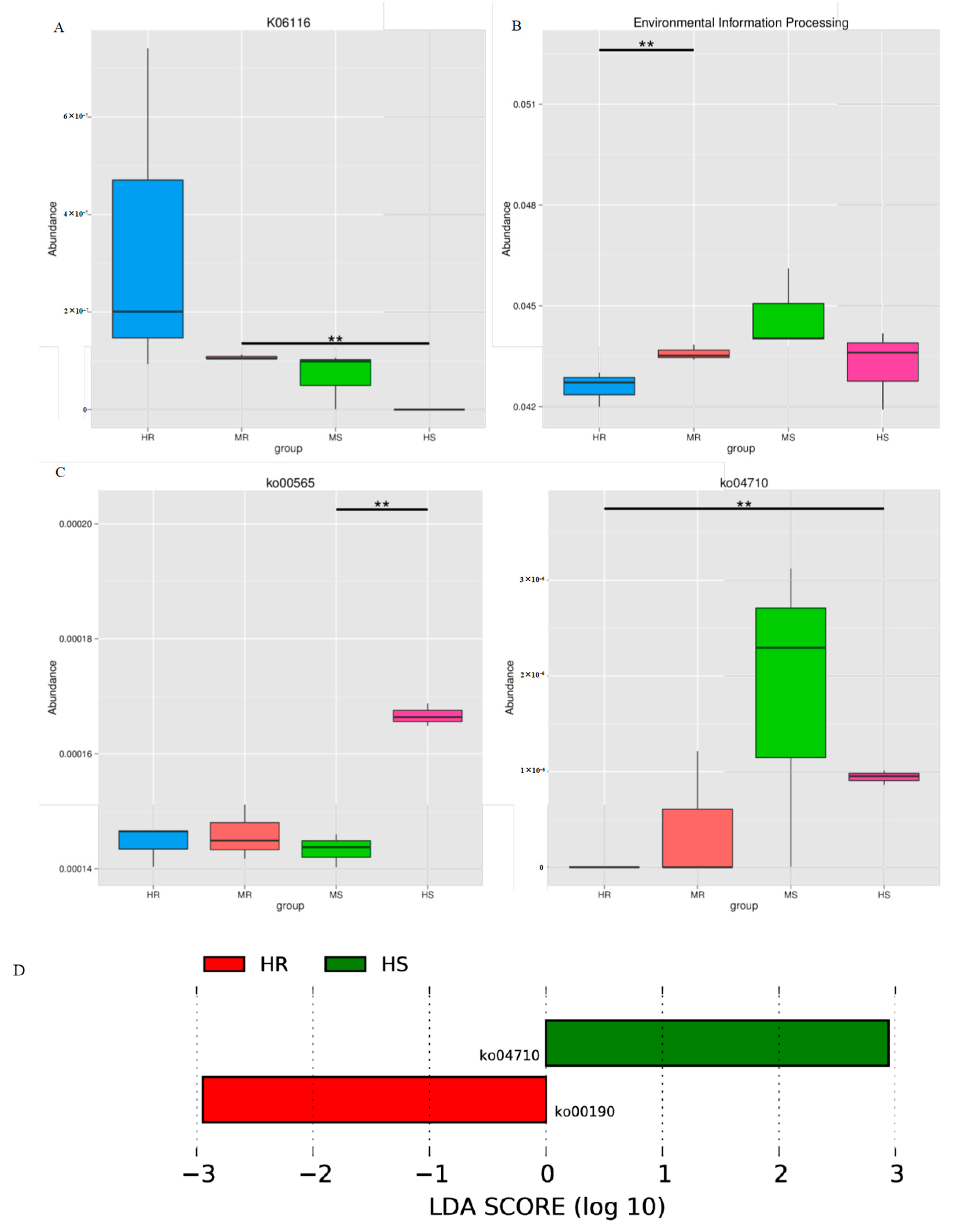
3.7. Representative Microbial Functions after Pathogen Infection
3.8. Analysis of Rhizosphere Microbial Antibiotic Resistance Ontologies (AROs)
4. Discussion
4.1. Response of Microbial Composition and Structure to Clubroot Disease
4.2. Differential Microbes and Their Relationship with Soil Physiochemical Properties
4.3. Response of Microbial Functional Adaptation to Clubroot Disease
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalloo, G.; Rana, M.K. 10-Chinese cabbage: Brassica pekinensis, B. chinensis. In Genetic Improvement of Vegetable Crops; Elsevier: Amsterdam, The Netherlands, 1993; pp. 179–186. [Google Scholar]
- Becke, S.; Friederike, M.D.; Christine, S. Risk potential of clubroot disease on winter oilseed rape. Plant Dis. 2015, 99, 667–675. [Google Scholar]
- Heo, S.H.; Jang, S.J.; Choi, J.S.; Jang, C.S.; Song, J.Y.; Kim, H.G. Chinese cabbage clubroot pathogen, Plasmodiophora brassicae, is genetically stable. Mycobiology 2009, 37, 225–229. [Google Scholar] [CrossRef]
- Hasan, J.; Megha, S.; Rahman, H. Clubroot in Brassica: Recent advances in genomics, breeding, and disease management. Genome 2021, 64, 735–760. [Google Scholar] [CrossRef]
- Han, X.M.; Cui, B.R.; Niu, Q.J. Research progress on resistance of Chinese cabbage to clubroot disease. Contemp. Hortic. 2022, 45, 87–89. (In Chinese) [Google Scholar]
- Peng, Y.L. Clubroot Distribution, Pathotype of Plasmodiophora brassicae in Sichuan and Clubroot Resistance. Ph.D. Thesis, Sichuan Agricultural University, Chengdu, China, 2018. (In Chinese). [Google Scholar]
- Hu, J.K.; Huang, R.; Tan, G.J.; Ding, Y.H.; Hua, J.L.; Li, X.S.; Bao, Z.M.; Huang, R.R. Comparison of control effects of different medicaments and disease-tolerant variety on clubroot disease of Chinese cabbage. Acta Agric. Jiangxi 2018, 30, 49–51+56. (In Chinese) [Google Scholar]
- Zhang, J.H.; Ahmed, W.; Zhou, X.H.; Yao, B.; He, Z.L.; Qiu, Y.; Wei, F.J.; He, Y.L.; Wei, L.F.; Ji, G.H. Crop rotation with marigold promotes soil bacterial structure to assist in mitigating clubroot Incidence in Chinese Cabbage. Plants 2022, 11, 2295. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Wang, P.F.; Zhang, Q.; Tian, T.; Li, Y.H.; Xu, J.Q.; Zhang, G.P.; Shi, J. Effect of biochar based amendment on soil aggregate structure and the growth of Chinese Cabbage (Brassica bara L.) with different resistance. J. Saf. Environ. 2022, 22, 1019–1026. [Google Scholar]
- Xie, G.L.; Zhang, Z.H.; Wu, L.T.; Liu, J.W.; Xu, Q.J.; Fu, K.J.; Nie, Q.; Zhang, J.L.; Lin, C.; Chen, W.H.; et al. Effect of biochar application combined with microbial agent on prevention and control of Plasmodiophora brassicae of Chinese cabbage. Southwest China J. Agric. Sci. 2023, 36, 105–111. (In Chinese) [Google Scholar]
- Ahmed, A.; Munir, S.; He, P.F.; Li, Y.M.; He, P.B.; Wu, Y.X.; He, Y.Q. Biocontrol arsenals of bacterial endophyte: An imminent triumph against clubroot disease. Microbiol. Res. 2020, 241, 126565. [Google Scholar] [CrossRef]
- Li, G.Q. Study on control techniques of clubroot disease of alpine Chinese cabbage in Lixian County, Sichuan Province. Ph.D. Thesis, Sichuan Agricultural University, Chengdu, China, 2018. (In Chinese). [Google Scholar]
- Gao, Y.H.; Zheng, Z.H.; Zhang, Y.; Hu, Y.G.; Wang, X.F. Mechanism of rhizosphere micro-ecology in controlling soil-borne fungal diseases: A review. J. China Agric. Univ. 2021, 26, 100–113. (In Chinese) [Google Scholar]
- He, P.J.; Cui, W.Y.; Shahzad, M.; He, P.B.; Huang, R.R.; Li, X.Y.; Wu, Y.X.; Wang, Y.H.; Yang, J.; Tang, P.; et al. Fengycin produced by Bacillus subtilis XF-1 plays a major role in the biocontrol of Chinese cabbage clubroot via direct effect and defense stimulation. J. Cell. Physiol. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Abdelaziz, A.M.; Hashem, A.H.; El-Sayyad, G.S.; El-Wakil, D.A.; Selim, S.; Alkhalifah, D.H.; Attia, M.S. Biocontrol of soil borne diseases by plant growth promoting rhizobacteria. Trop. Plant Pathol. 2023, 48, 105–127. [Google Scholar] [CrossRef]
- Raaijmakers, J.M.; Paulitz, T.C.; Steinberg, C.; Alabouvette, C.; Moënne-Loccoz, Y. The rhizosphere: A playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 2009, 321, 341–361. [Google Scholar] [CrossRef]
- Zhang, J.H.; Wei, L.F.; Yang, J.; Ahmed, W.; Wang, Y.T.; Fu, L.N.; Ji, G.H. Probiotic consortia: Reshaping the rhizospheric microbiome and its role in suppressing root-rot disease of panax notoginseng. Front. Microbiol. 2020, 11, 701. [Google Scholar] [CrossRef]
- Yang, S.D.; Liu, H.W.; Xie, P.H.; Wen, T.; Shen, Q.R.; Yuan, J. Emerging pathways for engineering the rhizosphere microbiome for optimal plant health. J. Agric. Food Chem. 2023, 71, 4441–4449. [Google Scholar] [CrossRef]
- Classen, A.T.; Sundqvist, M.K.; Henning, J.A.; Newman, G.S.; Moore, J.A.M.; Cregger, M.A.; Moorhead, L.C.; Patterson, C.M. Direct and indirect effects of climate change on soil microbial and soil microbial-plant interactions: What lies ahead? Ecosphere 2015, 6, art130. [Google Scholar] [CrossRef]
- Hu, R.; Zheng, L.; Liu, H.; Huang, J.B. Effects of straw returning on microbial diversity in rice rhizosphere and occurrence of rice sheath blight. Acta Phytophylacica Sin. 2020, 47, 1261–1269. (In Chinese) [Google Scholar]
- Deng, X.H.; Zhang, N.; Li, Y.C.; Zhu, C.Z.; Qu, B.Y.; Liu, H.J.; Li, R.; Bai, Y.; Shen, Q.R.; Joana, F.S. Bio-organic soil amendment promotes the suppression of Ralstonia solanacearum by inducing changes in the functionality and composition of rhizosphere bacterial communities. New Phytol. 2022, 235, 1558–1574. [Google Scholar] [CrossRef]
- Zhu, F.Y.; Fang, Y.; Wang, Z.W.; Wang, P.; Yang, K.K.; Xiao, L.T.; Wang, R.Z. Salicylic acid remodeling of the rhizosphere microbiome induces watermelon root resistance against Fusarium oxysporum f. sp. niveum infection. Front. Microbiol. 2022, 13, 1015038. [Google Scholar] [CrossRef]
- Sudini, H.; Liles, M.R.; Arias, C.R.; Bowen, K.L.; Huettel, R.N. Exploring soil bacterial communities in different peanut-cropping sequences using multiple molecular approaches. Phytopathology 2011, 101, 819–827. [Google Scholar] [CrossRef]
- Yang, X.X.; Huang, X.Q.; Wu, W.X.; Xiang, Y.J.; Du, L.; Zhang, L.; Liu, Y. Effects of different rotation patterns on the occurrence of clubroot disease and diversity of rhizosphere microbes. J. Integr. Agric. 2020, 19, 2265–2273. [Google Scholar] [CrossRef]
- Cai, Q.H.; Zhou, G.S.; Ahmed, W.; Cao, Y.Y.; Zhao, M.W.; Li, Z.H.; Zhao, Z.X. Study on the relationship between bacterial wilt and rhizospheric microbial diversity of flue-cured tobacco cultivars. Eur. J. Plant Pathol. 2021, 160, 265–276. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.M.; Singh, B.K. Plant-microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.T.; Liu, F.; Liang, J.M.; Zhao, P.; Tsui, C.K.M.; Cai, L. Cross-kingdom synthetic microbiota supports tomato suppression of Fusarium wilt disease. Nat. Commun. 2022, 13, 7890. [Google Scholar] [CrossRef]
- Zhou, X.G.; Zhang, J.Y.; u Rahman, M.K.; Gao, D.M.; Wei, Z.; Wu, F.Z.; Francisco, D.A. Interspecific plant interaction via root exudates structures the disease suppressiveness of rhizosphere microbiomes. Mol. Plant 2023, 16, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.W.; Brettell, L.E.; Qiu, Z.G.; Singh, B.K. Microbiome-mediated stress resistance in plants. Trends Plant Sci. 2020, 25, 733–743. [Google Scholar] [CrossRef]
- Alicia, B.S.; Samuel, J.; Deetja, R.A.; Liu, B.Y.; Flachowsky, H.; Winkelmann, T.; Beerhues, L.; Nesme, J.; Søren, J.S.; Doris, V.; et al. Root exposure to apple replant disease soil triggers local defense response and rhizoplane microbiome dysbiosis. FEMS Microbiol. Ecol. 2021, 97, fiab031. [Google Scholar]
- Chen, H.; Ren, H.Y.; Liu, J.J.; Tian, Y.; Lu, S.G. Soil acidification induced decline disease of Myrica rubra: Aluminum toxicity and bacterial community response analyses. Environ. Sci. Pollut. Res. 2022, 29, 45435–45448. [Google Scholar] [CrossRef]
- Wang, B.; Wang, X.C.; Wang, Z.W.; Zhu, K.F.; Wu, W.M. Comparative metagenomic analysis reveals rhizosphere microbial community composition and functions help protect grapevines against salt stress. Front. Microbiol. 2023, 14, 1102547. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.B.; Dawn, C.; Grady, P.; Deng, S.W.; Gao, C.; Spencer, D.; Simmons, T.; Wipf, H.M.L.; Daniel, C.; et al. Genome-resolved metagenomics reveals role of iron metabolism in drought-induced rhizosphere microbiome dynamics. Nat. Commun. 2021, 12, 3209. [Google Scholar] [CrossRef]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef]
- Lu, P.; Shi, H.L.; Tao, J.M.; Jin, J.J.; Wang, S.J.; Zheng, Q.X.; Liu, P.P.; Xiang, B.K.; Chen, Q.S.; Xu, Y.L.; et al. Metagenomic insights into the changes in the rhizosphere microbial community caused by the root-knot nematode Meloidogyne incognita in tobacco. Environ. Res. 2023, 216, 114848. [Google Scholar] [CrossRef]
- Liu, C.M.; Yang, Z.F.; He, P.F.; Munir, S.; Wu, Y.X.; Ho, H.; He, Y.Q. Deciphering the bacterial and fungal communities in clubroot-affected cabbage rhizosphere treated with Bacillus subtilis XF-1. Agric. Ecosyst. Environ. 2018, 256, 12–22. [Google Scholar] [CrossRef]
- Park, J.S.; Park, J.H.; Park, Y.D. Construction of pseudomolecule sequences of Brassica rapa ssp. pekinensis inbred line CT001 and analysis of spontaneous mu-tations derived via sexual propagation. PLoS ONE 2019, 14, e0222283. [Google Scholar]
- Langdon, W.B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Mining 2015, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Oh, J.; Byrd, A.L.; Deming, C.; Conlan, S.; Kong, H.H.; Segre, J.A. Biogeography and individuality shape function in the human skin metagenome. Nature 2014, 514, 59–64. [Google Scholar] [CrossRef]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Nagarajan, N.; Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef]
- He, P.J.; Cui, W.Y.; Mnuir, S.; He, P.F.; Li, X.Y.; Wu, Y.X.; Yang, X.W.; Tang, P.; He, Y.Q. Plasmodiophora brassicae root hair interaction and control by Bacillus subtilis XF-1 in Chinese cabbage. Biol. Control. 2019, 128, 56–63. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Deng, Y.S.; Nie, Q.; Xie, G.L.; Wu, L.T.; Di, X.Y.; Shi, H.; Fu, K.J.; Zhang, J.L.; Lin, C.; et al. Differences on soil microbial community and function of healthy and clubroot diseased plants of Chinese cabbage. Chin. J. Eco-Agric. 2023, 31, 530–542. [Google Scholar]
- Obieze, C.C.; George, P.B.L.; Boyle, B.; Khasa, D.P. Black pepper rhizomicrobiome: Spectrum of plant health indicators, critical environmental factors and community compartmentation in Vietnam. Appl. Soil Ecol. 2023, 187, 104857. [Google Scholar] [CrossRef]
- Wen, T.; Ding, Z.X.; Thomashow, L.S.; Hale, L.; Yang, S.D.; Xie, P.H.; Liu, X.Y.; Wang, H.Q.; Shen, Q.R.; Yuan, J. Deciphering the mechanism of fungal pathogen-induced disease-suppressive soil. New Phytol. 2023, 238, 2634–2650. [Google Scholar] [CrossRef] [PubMed]
- Tao, C.; Li, R.; Xiong, W.; Shen, Z.Z.; Kowalchuk, G.A. Bio-organic fertilizers stimulate indigenous soil Pseudomonas populations to enhance plant disease suppression. Microbiome 2020, 8, 137. [Google Scholar] [CrossRef]
- Wu, W.X.; Huang, X.Q.; Zhang, L.; Yang, X.X.; Li, H.Z.; Liu, Y. Crucifer clubroot disease changes the microbial community structure of rhizosphere soil. Acta Ecol. Sin. 2020, 40, 1532–1541. [Google Scholar]
- Hu, J.; Wei, Z.; Friman, V.P.; Gu, S.H.; Wang, X.F.; Eisenhauer, N.; Yang, T.J.; Ma, J.; Shen, Q.R.; Xu, Y.C.; et al. Probiotic diversity enhances rhizosphere microbiome function and plant disease suppression. MBio 2017, 7, e01790-16. [Google Scholar] [CrossRef]
- Hu, Y.L.; Qiu, L.; Zhang, Z.J.; Liu, K.; Xia, X.; Xiong, S.L.; Zhao, S.M.; Zhao, Z.Q.; Hu, Y.M.; Liang, Y.X. Control of Streptomyces alfalfae XY25T over clubroot disease and its effect on rhizosphere microbial community in Chinese cabbage field trials. Front. Microbiol. 2021, 12, 641556. [Google Scholar] [CrossRef]
- Larkin, R.P.; Fravel, D.R. Effects of varying environmental conditions on biological control of Fusarium wilt of tomato by nonpathogenic Fusarium spp. Phytopathology 2002, 92, 1160–1166. [Google Scholar] [CrossRef]
- Zhou, J.H.; Yang, J.J.; Hui, K.; Feng, Z.Y.; Zhang, L.X.; Meng, C.F.; Guo, Q.; Lai, H.X. Effects of 3 biocontrol strains on clubroot disease-resistant and mechanism in Chinese cabbage. Acta Agric. Boreali-Occident. Sin. 2020, 29, 641–651. [Google Scholar]
- Kang, H.H.; Chai, A.L.; Lin, Z.H.; Shi, Y.X.; Xie, X.W.; Li, L.; Fan, T.F.; Xiang, S.; Xie, J.M.; Li, B.J. Deciphering differences in microbial community diversity between clubroot-diseased and healthy soils. Microorganisms 2024, 12, 251. [Google Scholar] [CrossRef]
- Mazoyon, C.; Hirel, B.; Pecourt, A.; Catterou, M.; Gutierrez, L.; Sarazin, V.; Dubois, F.; Duclercq, J. Sphingomonas sediminicola is an endosymbiotic bacterium able to induce the formation of root nodules in pea (Pisum sativum L.) and to enhance plant biomass production. Microorganisms 2023, 11, 199. [Google Scholar] [CrossRef]
- Jiang, H.; Peng, Y.M.; Yan, Y.F.; Zhang, Y.; Wu, C.J.; He, B.H.; Wang, X.Y. Screening, identification, and evaluation of antagonistic bacteria against clubroot of mustard. Plant Prot. 2018, 44, 104–110. [Google Scholar]
- Raio, A.; Puopolo, G. Pseudomonas chlororaphis metabolites as biocontrol promoters of plant health and improved crop yield. World J. Microbiol. Biotechnol. 2021, 37, 99. [Google Scholar] [CrossRef]
- Wang, X.H.; Ji, C.; Song, X.; Liu, Z.Y.; Liu, Y.; Gao, Q.X.; Li, C.H.; Zheng, R.; Han, X.H.; Liu, X.L. Biocontrol of two bacterial inoculant strains and their effects on the rhizosphere microbial community of field-grown wheat. BioMed Res. Int. 2021, 2021, 8835275. [Google Scholar] [CrossRef]
- Narisawa, K.; Shimura, M.; Usuki, F.; Fukuhara, S.; Hashiba, T. Effects of pathogen density, soil moisture, and soil pH on biological control of clubroot in Chinese cabbage by Heteroconium chaetospira. Plant Dis. 2005, 89, 285–290. [Google Scholar] [CrossRef]
- Zhang, S.N.; Wang, Y.; Sun, L.T.; Qiu, C.; Ding, Y.Q.; Gu, H.L.; Wang, L.J.; Wang, Z.S.; Ding, Z.T. Organic mulching positively regulates the soil microbial communities and ecosystem functions in tea plantation. BMC Microbiol. 2020, 20, 103. [Google Scholar] [CrossRef]
- Rashid, A.; Ahmed, H.U.; Xiao, Q.; Hwang, S.F.; Strelkov, S.E. Effects of root exudates and pH on Plasmodiophora brassicae resting spore germination and infection of canola (Brassica napus L.) root hairs. Crop Prot. 2013, 48, 16–23. [Google Scholar] [CrossRef]
- Hong, S.; Yuan, X.F.; Yang, J.M.; Yang, Y.; Jv, H.L.; Li, R.; Jia, Z.J.; Ruan, Y.Z. Selection of rhizosphere communities of diverse rotation crops reveals unique core microbiome associated with reduced banana Fusarium wilt disease. New Phytol. 2023, 238, 2194–2209. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Xiong, C.; Gao, C.; Tsui, C.K.M.; Wang, M.M.; Zhou, X.; Zhang, A.M.; Cai, L. Disease-induced changes in plant microbiome assembly and functional adaptation. Microbiome 2021, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Y.; Wang, F.; Zhang, H.Q.; Qin, F.J.; Xiang, W.; Wu, C.F.; Yan, C.Q.; Zhu, Z.K.; Chen, J.P.; Ge, T.D. Deciphering differences in microbial community composition and multifunctionality between healthy and Alternaria solani-infected potato rhizosphere soils. Plant Soil 2022, 484, 347–362. [Google Scholar] [CrossRef]
- Lu, N. Study of Salt Tolerance Mechanism in Aureobasidium pullulans. Master’s Thesis, Shenyang Agricultural University, Shenyang, China, 2020. [Google Scholar]
- Zhang, Q.C.; Li, W.H.; Han, X.D.; Wu, B.; Song, Z.Y.; Shi, J.Y. Plant glycerol suppresses brown rot of peach fruit by enhancing disease resistance. Physiol. 2024, 129, 102204. [Google Scholar] [CrossRef]
- Kátia, J.; Luís, F.M. Ketogenesis favors oxidative phosphorylation to promote disease tolerance. Trends Endocrinol. Metab. TEM 2024, 35, 177–179. [Google Scholar] [CrossRef]
- Martínez, J.L.; Coque, T.M.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 2014, 13, 116–123. [Google Scholar] [CrossRef]
- Thomas, C.A.H.; Ben, J.T.; Jessica, C.A.F.; Helen, O.; Christopher, J.C. Deep sequence analysis reveals the ovine rumen as a reservoir of antibiotic resistance genes. Environ. Pollut. 2018, 235, 571–575. [Google Scholar]
- Ma, L.P.; Xia, Y.; Li, B.; Yang, Y.; Li, L.G.; Tiedje, J.M.; Zhang, T. Metagenomic assembly reveals hosts of antibiotic resistance genes and the shared resistome in pig, chicken, and human Feces. Environ. Sci. Technol. 2016, 50, 420–427. [Google Scholar] [CrossRef]
- Li, C.W.; Zhang, L.; Zhang, Z.Z.; Zhang, G.Q.; Wei, R.P.; Wu, D.F.; Zhai, C.M.; Li, F.; Jin, S.Y.; Dong, C.; et al. Study on the vagial flora of giant pandas by using metagenomics. J. Sichuan Univ. (Nat. Sci. Ed.) 2020, 57, 612–620. [Google Scholar]
- Imperiali, N.; Dennert, F.; Schneider, J.; Laessle, T.; Velatta, C.; Fesselet, M.; Wyler, M.; Mascher, F.; Mavrodi, O.; Mavrodi, D.; et al. Relationships between root pathogen resistance, abundance and expression of Pseudomonas antimicrobial genes, and soil properties in representative Swiss agricultural soils. Front. Plant Sci. 2017, 8, 427. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Lai, J.; Sun, X.; Huang, L.; Sheng, Y.; Zhang, Q.; Zeng, H.; Zhang, Y.; Ye, P.; Wei, S. Comparative Metagenomic Analysis Reveals Rhizosphere Microbiome Assembly and Functional Adaptation Changes Caused by Clubroot Disease in Chinese Cabbage. Microorganisms 2024, 12, 1370. https://doi.org/10.3390/microorganisms12071370
Liu Y, Lai J, Sun X, Huang L, Sheng Y, Zhang Q, Zeng H, Zhang Y, Ye P, Wei S. Comparative Metagenomic Analysis Reveals Rhizosphere Microbiome Assembly and Functional Adaptation Changes Caused by Clubroot Disease in Chinese Cabbage. Microorganisms. 2024; 12(7):1370. https://doi.org/10.3390/microorganisms12071370
Chicago/Turabian StyleLiu, Yong, Jia Lai, Xiaofang Sun, Ling Huang, Yuzhen Sheng, Qianfang Zhang, Hualan Zeng, Yinchao Zhang, Pengsheng Ye, and Shugu Wei. 2024. "Comparative Metagenomic Analysis Reveals Rhizosphere Microbiome Assembly and Functional Adaptation Changes Caused by Clubroot Disease in Chinese Cabbage" Microorganisms 12, no. 7: 1370. https://doi.org/10.3390/microorganisms12071370