
Influence of Mycobiota in the Nasopharyngeal Tract of COVID-19 Patients
 , , , , , , , , , and
, , , , , , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. COVID-19 Patients
2.2. Total RNA Extraction and Quality Checks
2.3. Library Preparation, Sequencing, and Bioinformatic Analyses
3. Results
3.1. Sequencing Quality
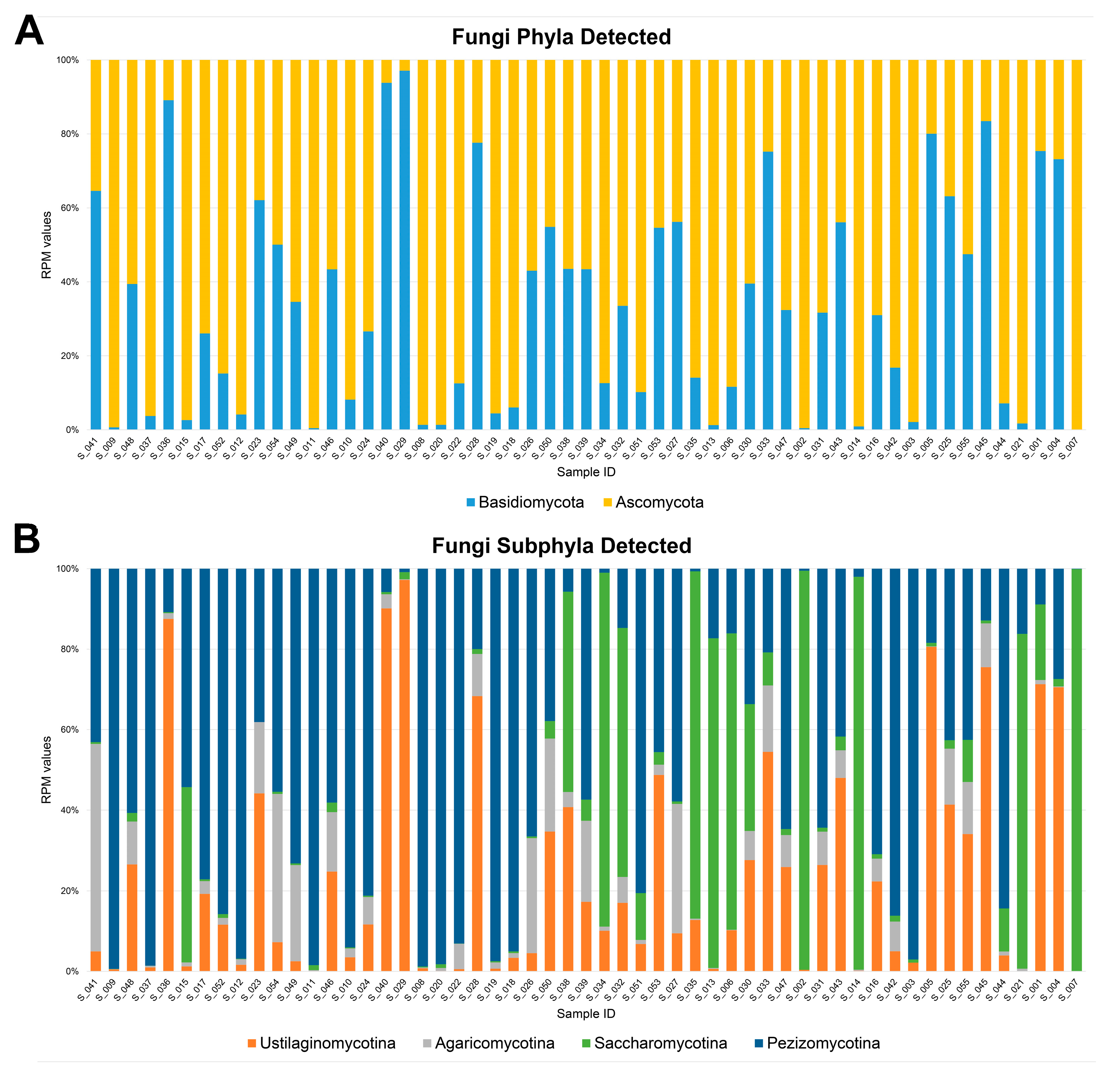
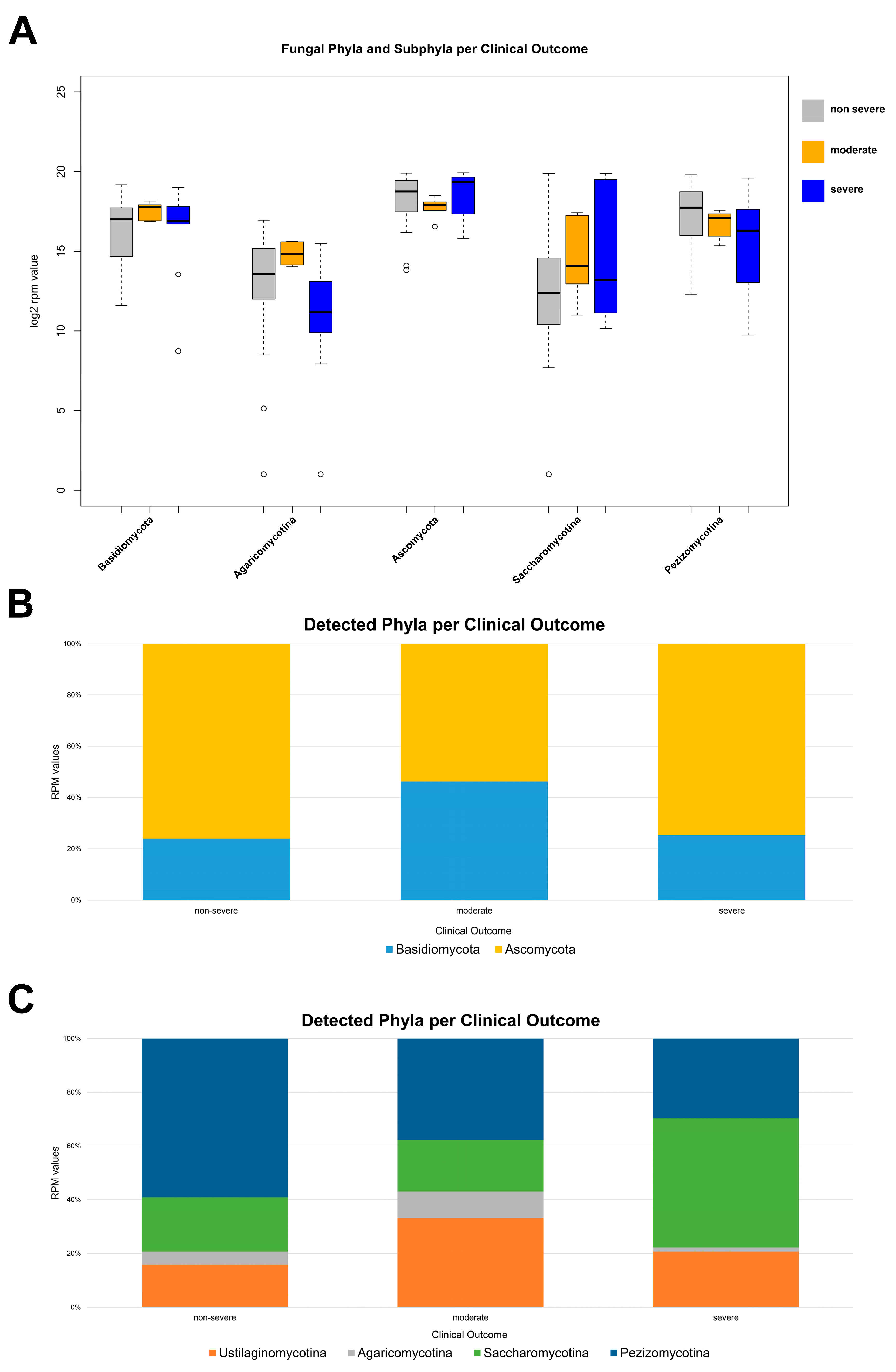
3.2. Fungal Diversity in the Nasal Cavity Varied with the Period and Severity of SARS-CoV-2 Infection
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between Microbiota and Immunity in Health and Disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Belvoncikova, P.; Splichalova, P.; Videnska, P.; Gardlik, R. The Human Mycobiome: Colonization, Composition and the Role in Health and Disease. J. Fungi 2022, 8, 1046. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Jin, L.; Lei, P.; Shao, C.; Wu, S.; Yang, Y.; He, Y.; Ren, R.; Xu, J. Whole-Genome Assembly and Analysis of a Medicinal Fungus: Inonotus Hispidus. Front. Microbiol. 2022, 13, 967135. [Google Scholar] [CrossRef] [PubMed]
- Prevel, R.; Boyer, P.; Beaufils, F.; Orieux, A.; Berger, P.; Boyer, A.; Delhaes, L.; Gruson, D. The Influence of Lung and Oropharyngeal Microbiota-Mycobiota on Ventilator-Associated Pneumonia Occurrence in Critically Ill Patients: A Systematic Review. Microbiota Health Dis. 2020, 2, e326. [Google Scholar] [CrossRef]
- Shajiei, A.; Liu, L.; Seinen, J.; Dieperink, W.; Hammerschmidt, S.; van Dijl, J.M.; Harmsen, H.J.M. Specific Associations between Fungi and Bacteria in Broncho-Alveolar Aspirates from Mechanically Ventilated Intensive Care Unit Patients. Virulence 2022, 13, 2022–2031. [Google Scholar] [CrossRef] [PubMed]
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation, and Treatment of Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Hernandez Acosta, R.A.; Esquer Garrigos, Z.; Marcelin, J.R.; Vijayvargiya, P. COVID-19 Pathogenesis and Clinical Manifestations. Infect. Dis. Clin. N. Am. 2022, 36, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.N.; Siefker, D.; Vu, L.; You, D.; DeVincenzo, J.; Pierre, J.F.; Cormier, S.A. Altered Gut Microbiota in Infants Is Associated with Respiratory Syncytial Virus Disease Severity. BMC Microbiol. 2020, 20, 140. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.J.; Mansbach, J.M.; Ajami, N.J.; Petrosino, J.F.; Zhu, Z.; Liang, L.; Camargo, C.A.; Hasegawa, K. Serum Metabolome Is Associated With the Nasopharyngeal Microbiota and Disease Severity Among Infants With Bronchiolitis. J. Infect. Dis. 2019, 219, 2005–2014. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.-Y.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut Microbiota Composition Reflects Disease Severity and Dysfunctional Immune Responses in Patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef]
- Soffritti, I.; D’Accolti, M.; Fabbri, C.; Passaro, A.; Manfredini, R.; Zuliani, G.; Libanore, M.; Franchi, M.; Contini, C.; Caselli, E. Oral Microbiome Dysbiosis Is Associated With Symptoms Severity and Local Immune/Inflammatory Response in COVID-19 Patients: A Cross-Sectional Study. Front. Microbiol. 2021, 12, 687513. [Google Scholar] [CrossRef]
- Giugliano, R.; Sellitto, A.; Ferravante, C.; Rocco, T.; D’Agostino, Y.; Alexandrova, E.; Lamberti, J.; Palumbo, D.; Galdiero, M.; Vaccaro, E.; et al. NGS Analysis of Nasopharyngeal Microbiota in SARS-CoV-2 Positive Patients during the First Year of the Pandemic in the Campania Region of Italy. Microb. Pathog. 2022, 165, 105506. [Google Scholar] [CrossRef] [PubMed]
- Ferravante, C.; Sanna, G.; Melone, V.; Fromentier, A.; Rocco, T.; D’Agostino, Y.; Lamberti, J.; Alexandrova, E.; Pecoraro, G.; Pagliano, P.; et al. Nasopharyngeal Virome Analysis of COVID-19 Patients during Three Different Waves in Campania Region of Italy. J. Med. Virol. 2022, 94, 2275–2283. [Google Scholar] [CrossRef] [PubMed]
- Davitt, E.; Davitt, C.; Mazer, M.B.; Areti, S.S.; Hotchkiss, R.S.; Remy, K.E. COVID-19 Disease and Immune Dysregulation. Best. Pract. Res. Clin. Haematol. 2022, 35, 101401. [Google Scholar] [CrossRef] [PubMed]
- Ferravante, C.; Arslan-Gatz, B.S.; Dell’Annunziata, F.; Palumbo, D.; Lamberti, J.; Alexandrova, E.; Di Rosa, D.; Strianese, O.; Giordano, A.; Palo, L.; et al. Dynamics of Nasopharyngeal Tract Phageome and Association with Disease Severity and Age of Patients during Three Waves of COVID-19. J. Med. Virol. 2022, 94, 5567–5573. [Google Scholar] [CrossRef]
- Ferravante, C.; Memoli, D.; Palumbo, D.; Ciaramella, P.; Di Loria, A.; D’Agostino, Y.; Nassa, G.; Rizzo, F.; Tarallo, R.; Weisz, A.; et al. HOME-BIO (SHOtgun MEtagenomic Analysis of BIOlogical Entities): A Specific and Comprehensive Pipeline for Metagenomic Shotgun Sequencing Data Analysis. BMC Bioinform. 2021, 22, 106. [Google Scholar] [CrossRef] [PubMed]
- Feldman, C.; Anderson, R. The Role of Co-Infections and Secondary Infections in Patients with COVID-19. Pneumonia (Nathan) 2021, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Candel, S.; Tyrkalska, S.D.; Álvarez-Santacruz, C.; Mulero, V. The Nasopharyngeal Microbiome in COVID-19. Emerg. Microbes Infect. 2023, 12, e2165970. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.F.; Marinkovic, A.; Prakash, S.; Sanyaolu, A.; Smith, S. COVID-19 and the Human Gut Microbiome: An Under-Recognized Association. Chonnam Med. J. 2022, 58, 96–101. [Google Scholar] [CrossRef]
- Merenstein, C.; Bushman, F.D.; Collman, R.G. Alterations in the Respiratory Tract Microbiome in COVID-19: Current Observations and Potential Significance. Microbiome 2022, 10, 165. [Google Scholar] [CrossRef]
- Zhang, J.L.; Si, H.F.; Shang, X.F.; Zhang, X.K.; Li, B.; Zhou, X.Z.; Zhang, J.Y. New Life for an Old Drug: In Vitro and in Vivo Effects of the Anthelmintic Drug Niclosamide against Toxoplasma Gondii RH Strain. Int. J. Parasitol. Drugs Drug Resist. 2019, 9, 27–34. [Google Scholar] [CrossRef]
- Zuo, T.; Zhan, H.; Zhang, F.; Liu, Q.; Tso, E.Y.K.; Lui, G.C.Y.; Chen, N.; Li, A.; Lu, W.; Chan, F.K.L.; et al. Alterations in Fecal Fungal Microbiome of Patients with COVID-19 During Time of Hospitalization until Discharge. Gastroenterology 2020, 159, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Motooka, D.; Kawasaki, T.; Oki, H.; Noda, Y.; Adachi, Y.; Niitsu, T.; Okamoto, S.; Tanaka, K.; Fukushima, K.; et al. Longitudinal Alterations of the Gut Mycobiota and Microbiota on COVID-19 Severity. BMC Infect. Dis. 2022, 22, 572. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.N.; Rahman, M.S.; Sarkar, M.M.H.; Habib, M.A.; Akter, S.; Banu, T.A.; Goswami, B.; Jahan, I.; Hossain, M.A.; Khan, M.S.; et al. Transcriptome Analysis Reveals Increased Abundance and Diversity of Opportunistic Fungal Pathogens in Nasopharyngeal Tract of COVID-19 Patients. PLoS ONE 2023, 18, e0278134. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xia, Y.; He, F.; Zhu, C.; Ren, W. Intestinal Mycobiota in Health and Diseases: From a Disrupted Equilibrium to Clinical Opportunities. Microbiome 2021, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Haegi, A.; Valente, M.T.; Riccioni, L.; Orzali, L.; Vitale, S.; Luongo, L.; Infantino, A. New-Generation Sequencing Technology in Diagnosis of Fungal Plant Pathogens: A Dream Comes True? J. Fungi 2022, 8, 737. [Google Scholar] [CrossRef] [PubMed]
- Reinold, J.; Farahpour, F.; Schoerding, A.-K.; Fehring, C.; Dolff, S.; Konik, M.; Korth, J.; van Baal, L.; Buer, J.; Witzke, O.; et al. The Fungal Gut Microbiome Exhibits Reduced Diversity and Increased Relative Abundance of Ascomycota in Severe COVID-19 Illness and Distinct Interconnected Communities in SARS-CoV-2 Positive Patients. Front. Cell Infect. Microbiol. 2022, 12, 848650. [Google Scholar] [CrossRef] [PubMed]
- Seyedjavadi, S.S.; Bagheri, P.; Nasiri, M.J.; Razzaghi-Abyaneh, M.; Goudarzi, M. Fungal Infection in Co-Infected Patients With COVID-19: An Overview of Case Reports/Case Series and Systematic Review. Front. Microbiol. 2022, 13, 888452. [Google Scholar] [CrossRef]
- Choi, H.M.; Moon, S.Y.; Yang, H.I.; Kim, K.S. Understanding Viral Infection Mechanisms and Patient Symptoms for the Development of COVID-19 Therapeutics. Int. J. Mol. Sci. 2021, 22, 1737. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| March–May 2020 | September–November 2020 | January–February 2021 | |
|---|---|---|---|
| Age | |||
| 8–40 | 6 | 7 | - |
| 41–59 | 3 | 10 | - |
| 60–69 | 5 | 4 | 4 |
| >70 | 8 | 4 | 1 |
| unknown | 3 | - | - |
| Gender | |||
| male | 15 | 17 | 4 |
| female | 8 | 8 | 1 |
| unknown | 2 | - | - |
| Disease severity | |||
| non-severe | 18 | 21 | - |
| moderate | 2 | - | 4 |
| severe | 5 | 4 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Folliero, V.; Ferravante, C.; Dell’Annunziata, F.; Brancaccio, R.N.; D’Agostino, Y.; Giurato, G.; Manente, R.; Terenzi, I.; Greco, R.; Boccia, G.; et al. Influence of Mycobiota in the Nasopharyngeal Tract of COVID-19 Patients. Microorganisms 2024, 12, 1468. https://doi.org/10.3390/microorganisms12071468
Folliero V, Ferravante C, Dell’Annunziata F, Brancaccio RN, D’Agostino Y, Giurato G, Manente R, Terenzi I, Greco R, Boccia G, et al. Influence of Mycobiota in the Nasopharyngeal Tract of COVID-19 Patients. Microorganisms. 2024; 12(7):1468. https://doi.org/10.3390/microorganisms12071468
Chicago/Turabian StyleFolliero, Veronica, Carlo Ferravante, Federica Dell’Annunziata, Rosario Nicola Brancaccio, Ylenia D’Agostino, Giorgio Giurato, Roberta Manente, Ilaria Terenzi, Rita Greco, Giovanni Boccia, and et al. 2024. "Influence of Mycobiota in the Nasopharyngeal Tract of COVID-19 Patients" Microorganisms 12, no. 7: 1468. https://doi.org/10.3390/microorganisms12071468