
Characterization of Mild Acid Stress Response in an Engineered Acid-Tolerant Escherichia coli Strain

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
2.2. RNA-Seq Quantification of Gene Expression Levels
2.3. Construction of the WCGNA Network
2.4. Hub Gene Determination
2.5. GO and KEGG Enrichment Analysis
2.6. PPI Analysis
2.7. Data Visualization
3. Results
3.1. Growth and Transcriptome Changes under Mild Acid Stress
3.2. Differential Gene Expression Analysis
3.3. WGCNA Analysis
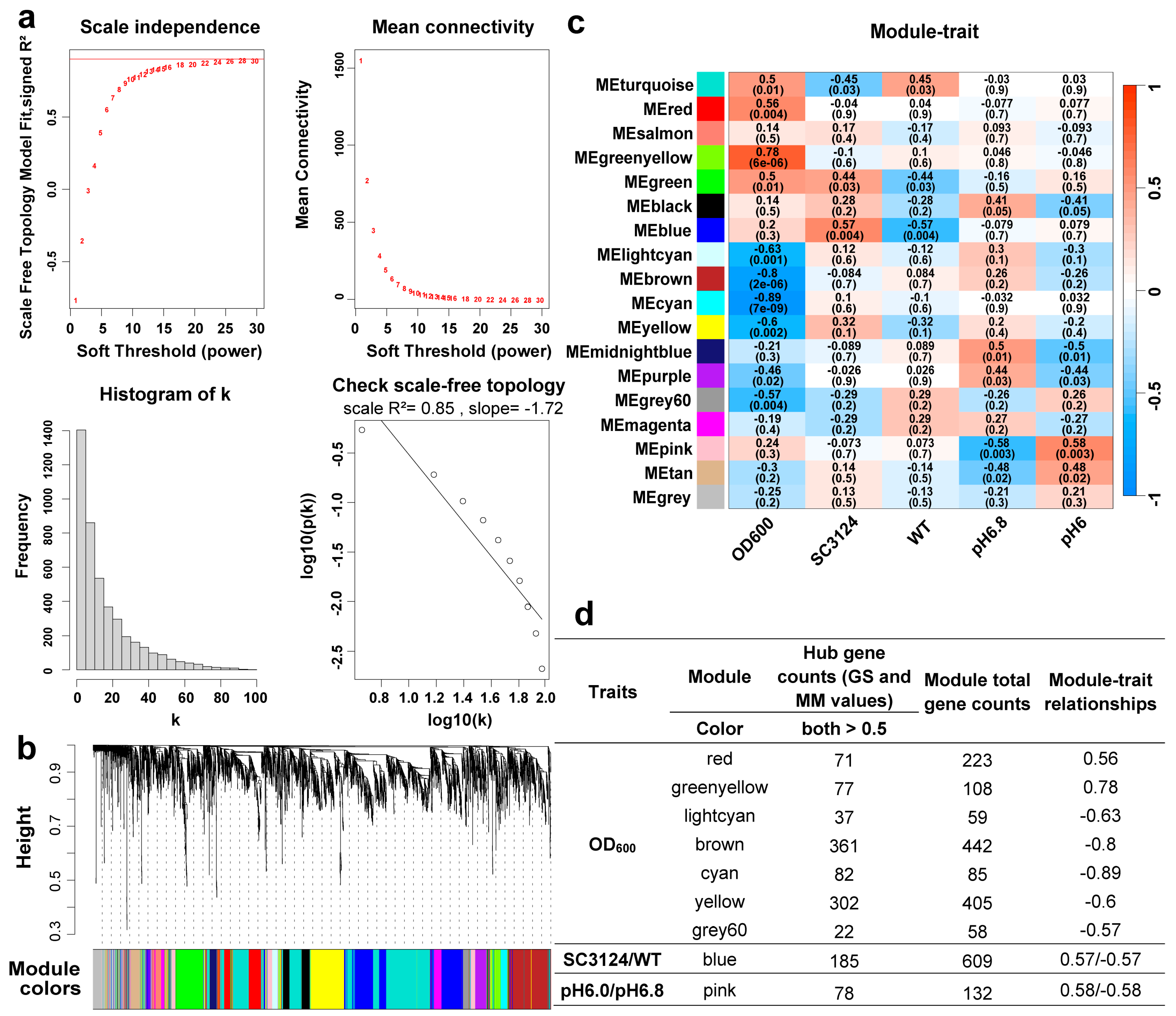
3.3.1. Soft Threshold Determination, Network Topology Analysis of Adjacency Matrices Based on WGCNA
3.3.2. Gene Clustering and Module–Trait Relationships
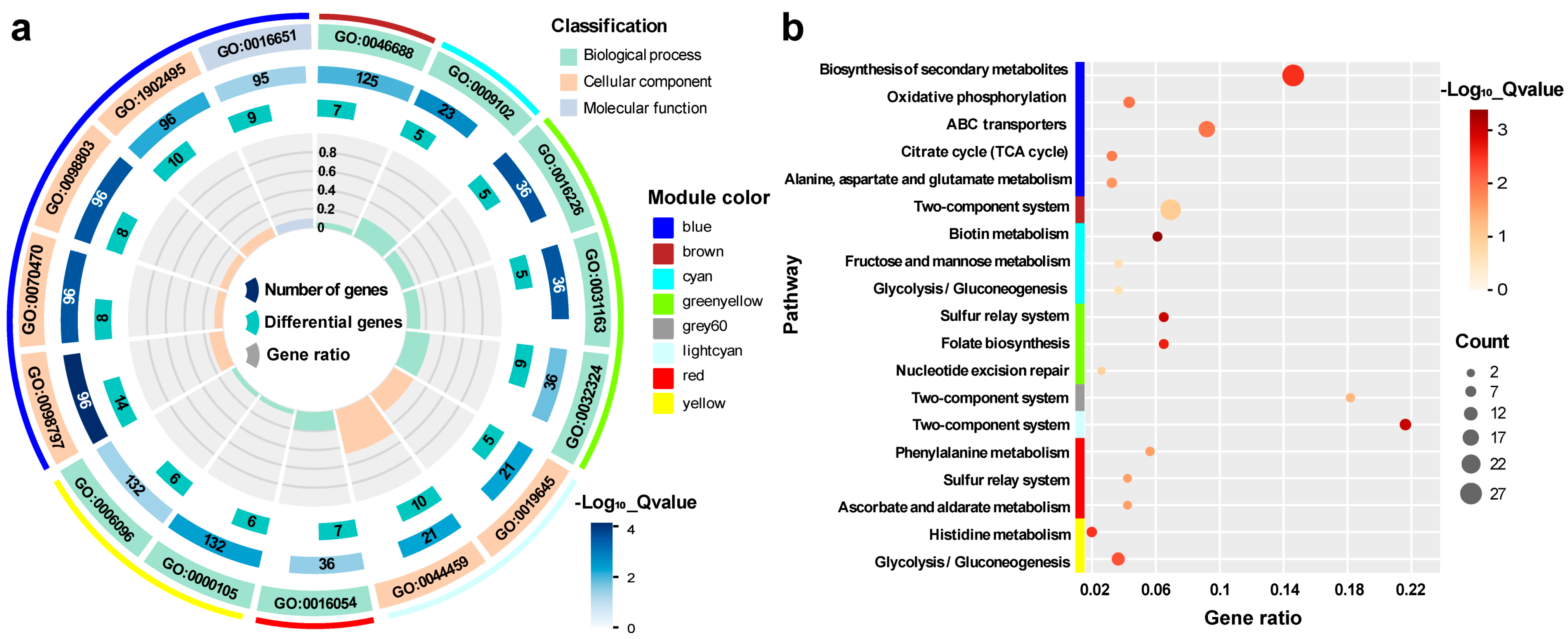
3.3.3. Functional Enrichment Analysis of Hub Genes Highly Correlated with Traits
3.4. OD600
3.5. SC3124 and WT
3.6. pH 6.0 and pH 6.8
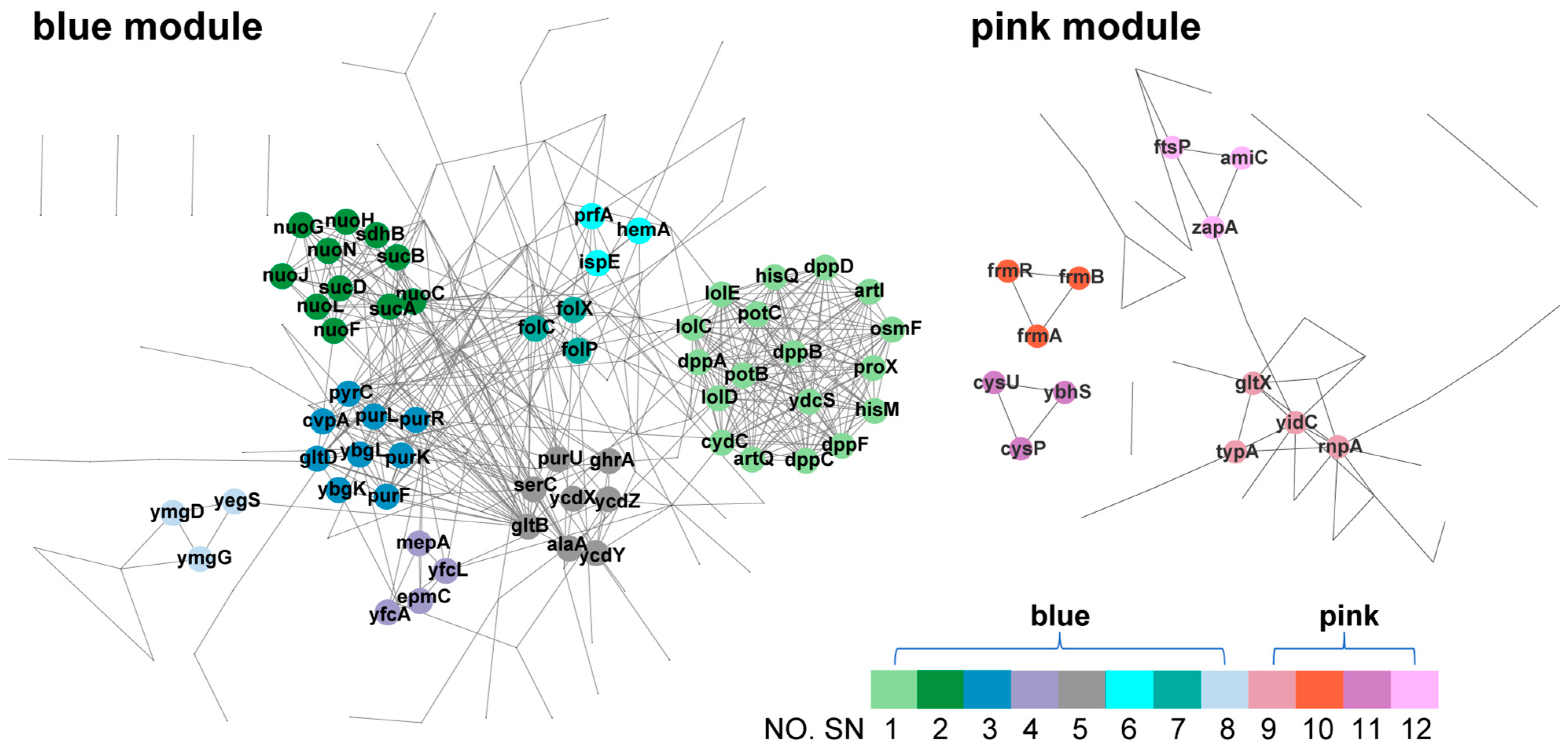
3.7. PPI Analysis of Hub Genes in Blue and Pink Modules
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foster, J.W. Escherichia coli Acid Resistance: Tales of an Amateur Acidophile. Nat. Rev. Microbiol. 2004, 2, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, K.; Ahmad, N.; Qin, L.; Li, C. Recent Advances in Engineering of Microbial Cell Factories for Intelligent Ph Regulation and Tolerance. Biotechnol. J. 2021, 16, e2100151. [Google Scholar] [CrossRef] [PubMed]
- Lund, P.; Tramonti, A.; De Biase, D. Coping with Low Ph: Molecular Strategies in Neutralophilic Bacteria. FEMS Microbiol. Rev. 2014, 38, 1091–1125. [Google Scholar] [CrossRef] [PubMed]
- Kanjee, U.; Houry, W.A. Mechanisms of Acid Resistance in Escherichia coli. Annu. Rev. Microbiol. 2013, 67, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Ledesma-Amaro, R.; Lu, M.; Jiang, Y.; Gao, S.; Jin, M.; Yang, S. Combined Evolutionary Engineering and Genetic Manipulation Improve Low Ph Tolerance and Butanol Production in a Synthetic Microbial Clostridium Community. Biotechnol. Bioeng. 2020, 117, 2008–2022. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yang, X.; Li, J.; Zhang, Y.; Chen, P.; Lin, Z. Engineered Global Regulator H-Ns Improves the Acid Tolerance of E. coli. Microb. Cell Factories 2018, 17, 118. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, J.; Yan, X.; Yang, J.; Li, X.; Chen, P.; Yang, X. Engineering of the Small Noncoding Rna (Srna) Dsra Together with the Srna Chaperone Hfq Enhances the Acid Tolerance of Escherichia coli. Appl. Environ. Microbiol. 2021, 87, e02923-20. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhang, W.; Chen, M.; Pan, J.; Lu, W.; Ping, S.; Yan, Y.; Hou, X.; Yuan, M.; Zhan, Y.; et al. Genome-Wide Transcriptome and Proteome Analysis of Escherichia coli Expressing Irre, a Global Regulator of Deinococcus Radiodurans. Mol. BioSyst. 2011, 7, 1613–1620. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Jiang, L.; Zhu, L.; Xu, Q.; Xu, X.; Huang, H. Tailoring of Global Transcription Sigma D Factor by Random Mutagenesis to Improve Escherichia coli Tolerance Towards Low-Phs. J. Biotechnol. 2016, 224, 55–63. [Google Scholar] [CrossRef]
- Jia, H.; Fan, Y.; Feng, X.; Li, C. Enhancing Stress-Resistance for Efficient Microbial Biotransformations by Synthetic Biology. Front. Bioeng. Biotechnol. 2014, 2, 44. [Google Scholar] [CrossRef]
- de Siqueira, G.M.V.; Silva-Rocha, R.; Guazzaroni, M.E. Turning the Screw: Engineering Extreme Ph Resistance in Escherichia coli through Combinatorial Synthetic Operons. ACS Synth. Biol. 2020, 9, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Yang, L.; Lloyd, C.J.; Fang, X.; Palsson, B.O. Genome-Scale Model of Metabolism and Gene Expression Provides a Multi-Scale Description of Acid Stress Responses in Escherichia coli. PLoS Comput. Biol. 2019, 15, e1007525. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wilson, J.; Ercanbrack, C.; Smith, H.; Gan, Q.; Fan, C. Genome-Wide Screening of Oxidizing Agent Resistance Genes in Escherichia coli. Antioxidants 2021, 10, 861. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Liu, P.; Chen, B.; Wang, X.; Tao, F.; Lin, Z.; Yang, X. Synthetic Acid Stress-Tolerance Modules Improve Growth Robustness and Lysine Productivity of Industrial Escherichia coli in Fermentation at Low Ph. Microb. Cell Factories 2022, 21, 68. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Gong, S.; Richard, H.; Tucker, D.L.; Conway, T.; Foster, J.W. Gade (Yhie) Activates Glutamate Decarboxylase-Dependent Acid Resistance in Escherichia coli K-12. Mol. Microbiol. 2003, 49, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Malki, A.; Le, H.T.; Milles, S.; Kern, R.; Caldas, T.; Abdallah, J.; Richarme, G. Solubilization of Protein Aggregates by the Acid Stress Chaperones Hdea and Hdeb. J. Biol. Chem. 2008, 283, 13679–13687. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.L.; Kim, I.G. Transcription of Ahpc, Katg, and Kate Genes in Escherichia coli Is Regulated by Polyamines: Polyamine-Deficient Mutant Sensitive to H2o2-Induced Oxidative Damage. Biochem. Biophys. Res. Commun. 2003, 301, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Ezraty, B.; Gennaris, A.; Barras, F.; Collet, J.F. Oxidative Stress, Protein Damage and Repair in Bacteria. Nat. Rev. Microbiol. 2017, 15, 385–396. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Wgcna: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Eigengene Networks for Studying the Relationships between Co-Expression Modules. BMC Syst. Biol. 2007, 1, 54. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. Htseq—A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for Rna-Seq Data with Deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Ren, R.; Liu, Z.; Song, M.; Li, J.; Wu, Z.; Ren, X.; Fu, L.; Li, W.; Zhang, W.; et al. Stabilizing Heterochromatin by Dgcr8 Alleviates Senescence and Osteoarthritis. Nat. Commun. 2019, 10, 3329. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, J.; Yuan, J.; Jiang, X.; Jiang, L.; Li, Z.; Yin, Z.; Du, Y.; Zhao, G.; Liu, B.; et al. Gene Coexpression Network Analysis Reveals a Novel Metabolic Mechanism of Clostridium Acetobutylicum Responding to Phenolic Inhibitors from Lignocellulosic Hydrolysates. Biotechnol. Biofuels 2020, 13, 163. [Google Scholar] [CrossRef] [PubMed]
- Azam, M.; Zhang, S.; Li, J.; Ahsan, M.; Agyenim-Boateng, K.G.; Qi, J.; Feng, Y.; Liu, Y.; Li, B.; Qiu, L.; et al. Identification of Hub Genes Regulating Isoflavone Accumulation in Soybean Seeds Via Gwas and Wgcna Approaches. Front. Plant Sci. 2023, 14, 1120498. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. Kobas-I: Intelligent Prioritization and Exploratory Visualization of Biological Functions for Gene Enrichment Analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. Tbtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The String Database in 2023: Protein-Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W. An Automated Method for Finding Molecular Complexes in Large Protein Interaction Networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.F.; Castellani, L.G.; Draghi, W.O.; Pérez-Giménez, J.; Torres Tejerizo, G.A.; Pistorio, M. Proteomic Analysis of Rhizobium Favelukesii Lpu83 in Response to Acid Stress. J. Proteome Res. 2019, 18, 3615–3629. [Google Scholar] [CrossRef]
- Hayes, E.T.; Wilks, J.C.; Sanfilippo, P.; Yohannes, E.; Tate, D.P.; Jones, B.D.; Radmacher, M.D.; BonDurant, S.S.; Slonczewski, J.L. Oxygen Limitation Modulates Ph Regulation of Catabolism and Hydrogenases, Multidrug Transporters, and Envelope Composition in Escherichia coli K-12. BMC Microbiol. 2006, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- Maurer, L.M.; Yohannes, E.; Bondurant, S.S.; Radmacher, M.; Slonczewski, J.L. Ph Regulates Genes for Flagellar Motility, Catabolism, and Oxidative Stress in Escherichia coli K-12. J. Bacteriol. 2005, 187, 304–319. [Google Scholar] [CrossRef]
- Higgins, C.F. Abc Transporters: Physiology, Structure and Mechanism—An Overview. Res. Microbiol. 2001, 152, 205–210. [Google Scholar] [CrossRef]
- Zhu, Z.; Yang, J.; Yang, P.; Wu, Z.; Zhang, J.; Du, G. Enhanced Acid-Stress Tolerance in Lactococcus Lactis Nz9000 by Overexpression of Abc Transporters. Microb. Cell Factories 2019, 18, 136. [Google Scholar] [CrossRef]
- Wang, G.; Li, D.; Ma, X.; An, H.; Zhai, Z.; Ren, F.; Hao, Y. Functional Role of Oppa Encoding an Oligopeptide-Binding Protein from Lactobacillus Salivarius Ren in Bile Tolerance. J. Ind. Microbiol. Biotechnol. 2015, 42, 1167–1174. [Google Scholar] [CrossRef]
- Madeo, M.; O’Riordan, N.; Fuchs, T.M.; Utratna, M.; Karatzas, K.A.; O’Byrne, C.P. Thiamine Plays a Critical Role in the Acid Tolerance of Listeria Monocytogenes. FEMS Microbiol. Lett. 2012, 326, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Lee, H.G.; Pi, K.; Choi, Y.J. The Effect of Low Ph on Protein Expression by the Probiotic Bacterium Lactobacillus Reuteri. Proteomics 2008, 8, 1624–1630. [Google Scholar] [CrossRef]
- Borezee, E.; Pellegrini, E.; Berche, P. Oppa of Listeria Monocytogenes, an Oligopeptide-Binding Protein Required for Bacterial Growth at Low Temperature and Involved in Intracellular Survival. Infect. Immun. 2000, 68, 7069–7077. [Google Scholar] [CrossRef]
- Sheehan, V.M.; Sleator, R.D.; Hill, C.; Fitzgerald, G.F. Improving Gastric Transit, Gastrointestinal Persistence and Therapeutic Efficacy of the Probiotic Strain Bifidobacterium Breve Ucc2003. Microbiology 2007, 153, 3563–3571. [Google Scholar] [CrossRef]
- Feehily, C.; Karatzas, K.A. Role of Glutamate Metabolism in Bacterial Responses Towards Acid and Other Stresses. J. Appl. Microbiol. 2013, 114, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Broadbent, J.R.; Larsen, R.L.; Deibel, V.; Steele, J.L. Physiological and Transcriptional Response of Lactobacillus Casei Atcc 334 to Acid Stress. J. Bacteriol. 2010, 192, 2445–2458. [Google Scholar] [CrossRef] [PubMed]
- El Rayes, J.; Rodríguez-Alonso, R.; Collet, J.F. Lipoproteins in Gram-Negative Bacteria: New Insights into Their Biogenesis, Subcellular Targeting and Functional Roles. Curr. Opin. Microbiol. 2021, 61, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Qin, Q.; Guo, H.; Liu, S.; Ge, S.; Zhang, H.; Cui, J.; Ren, F. Effect of Pre-Stressing on the Acid-Stress Response in Bifidobacterium Revealed Using Proteomic and Physiological Approaches. PLoS ONE 2015, 10, e0117702. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pang, X.; Wu, Y.; Wu, Y.; Xu, L.; Chen, Q.; Niu, J.; Zhang, X. New Insights into the Lactic Acid Resistance Determinants of Listeria Monocytogenes Based on Transposon Sequencing and Transcriptome Sequencing Analyses. Microbiol. Spectr. 2023, 11, e0275022. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Sakamoto, A.; Terui, Y.; Takao, K.; Sugita, Y.; Yamamoto, K.; Ishihama, A.; Igarashi, K.; Kashiwagi, K. Effect of Spermidine Analogues on Cell Growth of Escherichia coli Polyamine Requiring Mutant Ma261. PLoS ONE 2016, 11, e0159494. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, A.G.; Nesterova, L.Y. Polyamines as Modulators of Gene Expression under Oxidative Stress in Escherichia coli. Biochem. Biokhimiia 2003, 68, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Heunis, T.; Deane, S.; Smit, S.; Dicks, L.M. Proteomic Profiling of the Acid Stress Response in Lactobacillus Plantarum 423. J. Proteome Res. 2014, 13, 4028–4039. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, Z.; Tong, W.; Ding, Y.; Liu, B.; Shi, Y.; Wang, J.; Sun, S.; Liu, M.; Wang, Y.; et al. An Acid-Tolerance Response System Protecting Exponentially Growing Escherichia coli. Nat. Commun. 2020, 11, 1496. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subnetwork | Gene | Product | GO Pathway 1 | KEGG Pathway 1 |
|---|---|---|---|---|
| SN.1 | dppF | dipeptide ABC transporter ATP binding subunit DppF | Transporter activity; transmembrane transporter activity; nitrogen compound transport; establishment of localization; amino acid transport; carboxylic acid transport; monoatomic cation transmembrane transport; monoatomic ion transmembrane transport. | ABC transporters |
| lolE | lipoprotein release complex—inner membrane subunit | |||
| lolD | lipoprotein release complex—ATP binding subunit | |||
| artQ | L-arginine ABC transporter membrane subunit ArtQ | |||
| dppD | dipeptide ABC transporter ATP binding subunit DppD | |||
| artI | putative ABC transporter periplasmic binding protein ArtI | |||
| proX | glycine betaine ABC transporter periplasmic binding protein ProX | |||
| cydC | glutathione/L-cysteine ABC exporter subunit CydC | |||
| ydcS | putative ABC transporter periplasmic binding protein/polyhydroxybutyrate synthase | |||
| dppB | dipeptide ABC transporter membrane subunit DppB | |||
| potC | spermidine preferential ABC transporter membrane subunit PotC | |||
| hisM | lysine, arginine, ornithine ABC transporter/histidine ABC transporter, membrane subunit HisM | |||
| lolC | lipoprotein release complex—inner membrane subunit | |||
| potB | spermidine preferential ABC transporter membrane subunit PotB | |||
| hisQ | lysine, arginine, ornithine ABC transporter/histidine ABC transporter, membrane subunit HisQ | |||
| osmF | glycine betaine ABC transporter periplasmic binding protein OsmF | |||
| dppC | dipeptide ABC transporter membrane subunit DppC | |||
| dppA | dipeptide ABC transporter periplasmic binding protein | |||
| SN.2 | nuoJ | NADH:quinone oxidoreductase subunit J | NAD(P)H dehydrogenase (quinone) activity; oxidoreductase activity, acting on NAD(P)H; electron transfer activity. | Oxidative phosphorylation; citrate cycle (TCA cycle). |
| sucD | succinyl-CoA synthetase subunit alpha | |||
| nuoC | NADH:quinone oxidoreductase subunit CD | |||
| nuoN | NADH:quinone oxidoreductase subunit N | |||
| nuoF | NADH:quinone oxidoreductase subunit F | |||
| sucB | dihydrolipoyltranssuccinylase | |||
| nuoH | NADH:quinone oxidoreductase subunit H | |||
| nuoG | NADH:quinone oxidoreductase subunit G | |||
| sucA | subunit of E1(0) component of 2-oxoglutarate dehydrogenase | |||
| nuoL | NADH:quinone oxidoreductase subunit L | |||
| sdhB | succinate:quinone oxidoreductase, iron-sulfur cluster binding protein | |||
| SN.3 | pxpC | 5-oxoprolinase component C | Purine biosynthesis; pyrimidine nucleotide biosynthesis; glutamate biosynthesis; 5-oxoprolinase (ATP-hydrolyzing) activity; toxin biosynthesis. | Purine, pyrimidine metabolism; glutathione metabolism; alanine, aspartate and glutamate metabolism. |
| purR | DNA-binding transcriptional repressor PurR | |||
| pyrC | dihydroorotase | |||
| purF | amidophosphoribosyltransferase | |||
| pxpA | 5-oxoprolinase component A | |||
| purK | 5-(carboxyamino)imidazole ribonucleotide synthase | |||
| purL | phosphoribosylformylglycinamide synthetase | |||
| cvpA | colicin V production protein | |||
| gltD | glutamate synthase subunit GltD | |||
| SN.4 | epmC | EF-P-Lys34 hydroxylase | Not available. | Not available. |
| yfcA | putative transporter YfcA | |||
| yfcL | PF08891 family protein YfcL | |||
| mepA | peptidoglycan DD-endopeptidase/peptidoglycan LD-endopeptidase | |||
| SN.5 | alaA | glutamate--pyruvate aminotransferase AlaA | L-alanine biosynthesis; glutamate biosynthesis; serine biosynthesis; purine nucleotide biosynthesis; plasma membrane. | Alanine, aspartate and glutamate metabolism; glycine, serine and threonine metabolism; purine metabolism. |
| ycdX | zinc-binding phosphatase YcdX | |||
| serC | phosphoserine/phosphohydroxythreonine aminotransferase | |||
| ghrA | glyoxylate/hydroxypyruvate reductase A | |||
| purU | formyltetrahydrofolate deformylase | |||
| ycdY | chaperone protein YcdY | |||
| gltB | glutamate synthase subunit GltB | |||
| ycdZ | inner membrane protein YcdZ | |||
| SN.6 | prfA | peptide chain release factor RF1 | Not available. | Not available. |
| ispE | 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase | |||
| hemA | glutamyl-tRNA reductase | |||
| SN.7 | folC | bifunctional folylpolyglutamate synthetase/dihydrofolate synthetase | Folic acid biosynthetic process. | Folate biosynthesis. |
| folP | dihydropteroate synthase | |||
| folX | dihydroneopterin triphosphate 2′-epimerase | |||
| SN.8 | yegS | lipid kinase YegS | Not available | Not available |
| ymgG | PF13488 family protein YmgG | |||
| ymgD | PF16456 family protein YmgD |
| Subnetwork | Gene | Product | GO Pathway 1 | KEGG Pathway 1 |
|---|---|---|---|---|
| SN.9 | yidC | membrane protein insertase YidC | Not available | Not available |
| gltX | glutamate-tRNA ligase | |||
| rnpA | RNase P protein component | |||
| bipA | 50S ribosomal subunit assembly factor BipA | |||
| SN.10 | frmR | DNA-binding transcriptional repressor FrmR | Formaldehyde catabolic process | Microbial metabolism in diverse environments |
| frmB | S-formylglutathione hydrolase FrmB | |||
| frmA | S-(hydroxymethyl)glutathione dehydrogenase | |||
| SN.11 | cysP | thiosulfate/sulfate ABC transporter periplasmic binding protein CysP | Thiosulfate/sulfate transporter; sulfur metabolism; xenobiotic transmembrane transporter | Sulfur metabolism; ABC transporters |
| cysU | sulfate/thiosulfate ABC transporter inner membrane subunit CysU | |||
| ybhS | probable multidrug ABC transporter permease | |||
| SN.12 | amiC | N-acetylmuramoyl-L-alanine amidase C | Not available | Cationic antimicrobial peptide (CAMP) resistance |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, J.; Guo, H.; Wu, X.; Ma, S.; Zhang, X.; Yang, X.; Liu, B.; Feng, L.; Liu, H.; Huang, D. Characterization of Mild Acid Stress Response in an Engineered Acid-Tolerant Escherichia coli Strain. Microorganisms 2024, 12, 1565. https://doi.org/10.3390/microorganisms12081565
Qin J, Guo H, Wu X, Ma S, Zhang X, Yang X, Liu B, Feng L, Liu H, Huang D. Characterization of Mild Acid Stress Response in an Engineered Acid-Tolerant Escherichia coli Strain. Microorganisms. 2024; 12(8):1565. https://doi.org/10.3390/microorganisms12081565
Chicago/Turabian StyleQin, Jingliang, Han Guo, Xiaoxue Wu, Shuai Ma, Xin Zhang, Xiaofeng Yang, Bin Liu, Lu Feng, Huanhuan Liu, and Di Huang. 2024. "Characterization of Mild Acid Stress Response in an Engineered Acid-Tolerant Escherichia coli Strain" Microorganisms 12, no. 8: 1565. https://doi.org/10.3390/microorganisms12081565
APA StyleQin, J., Guo, H., Wu, X., Ma, S., Zhang, X., Yang, X., Liu, B., Feng, L., Liu, H., & Huang, D. (2024). Characterization of Mild Acid Stress Response in an Engineered Acid-Tolerant Escherichia coli Strain. Microorganisms, 12(8), 1565. https://doi.org/10.3390/microorganisms12081565