

New Iflavirus Species Characterized from Mosquitoes Captured in the Sao Paulo Zoological Facilities

 ,
,  , , , , ,
, , , , ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mosquito Collection
2.2. Identification of Mosquito Species
2.3. Sequencing
2.4. Bioinformatics and Phylogenetic Analysis
2.5. Genomic Annotation
2.6. Probability Mapping
2.7. Genetic Distance
3. Results and Discussion
3.1. Characterization of Mosquito Sampling and Genus
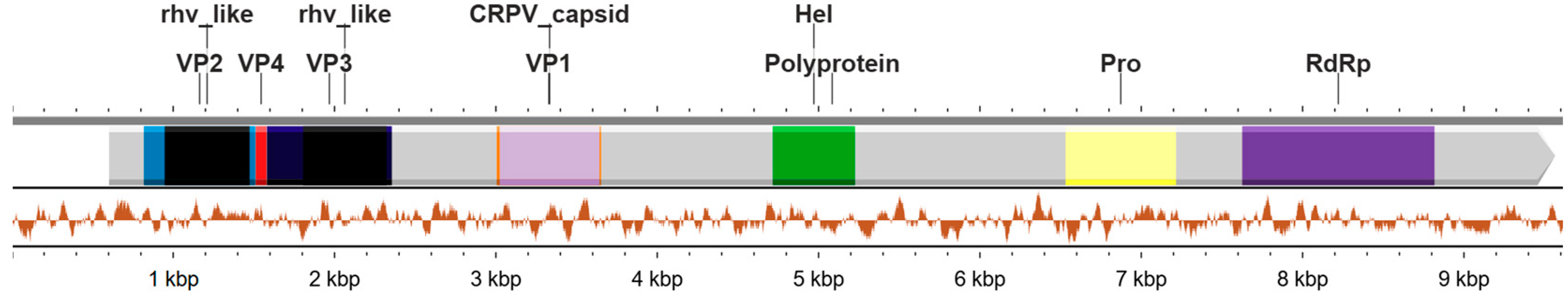
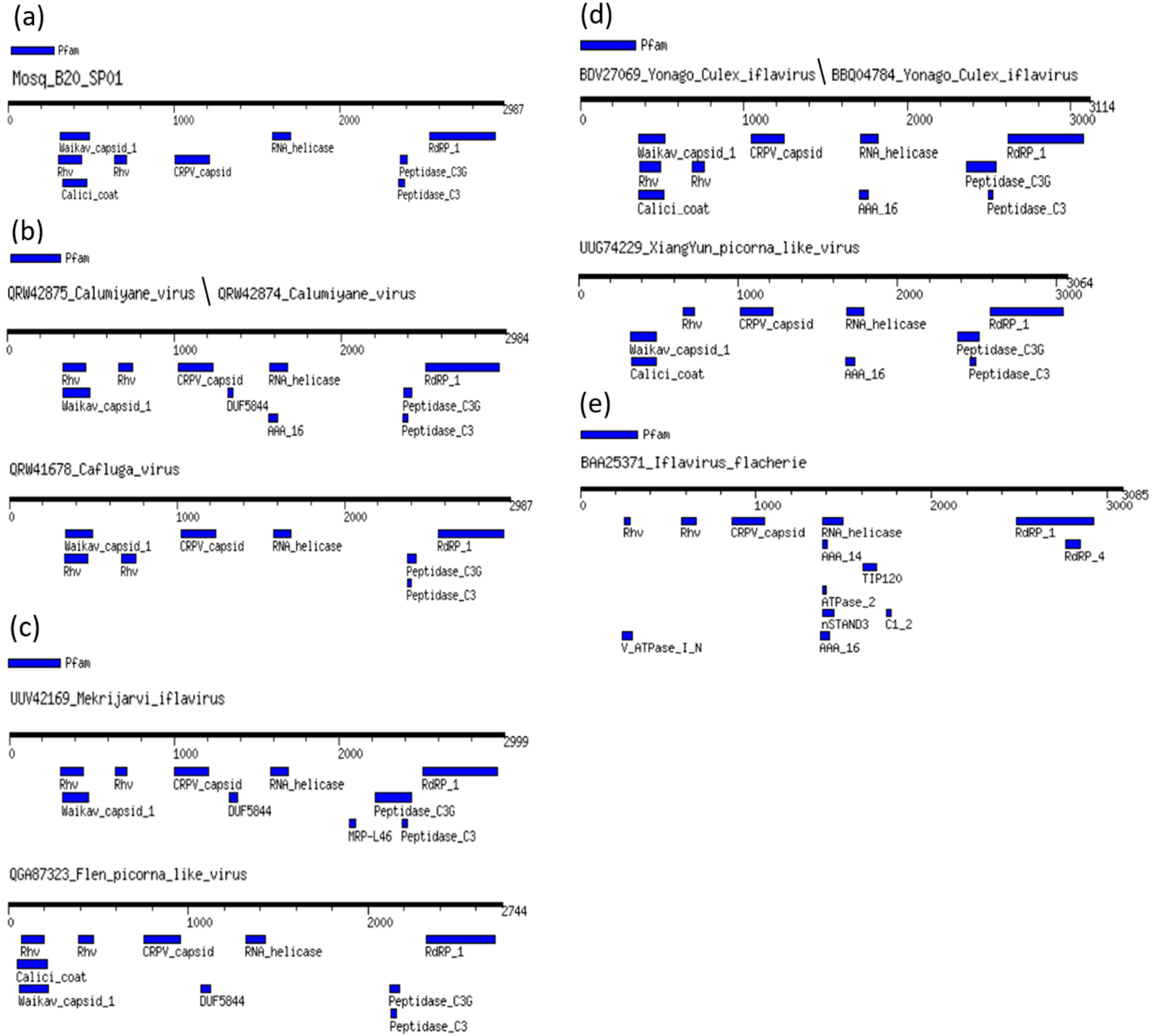
3.2. Genome Annotation of Mosq_B20_SP01 Identified in Anopheles
3.3. Phylogenetic Trees
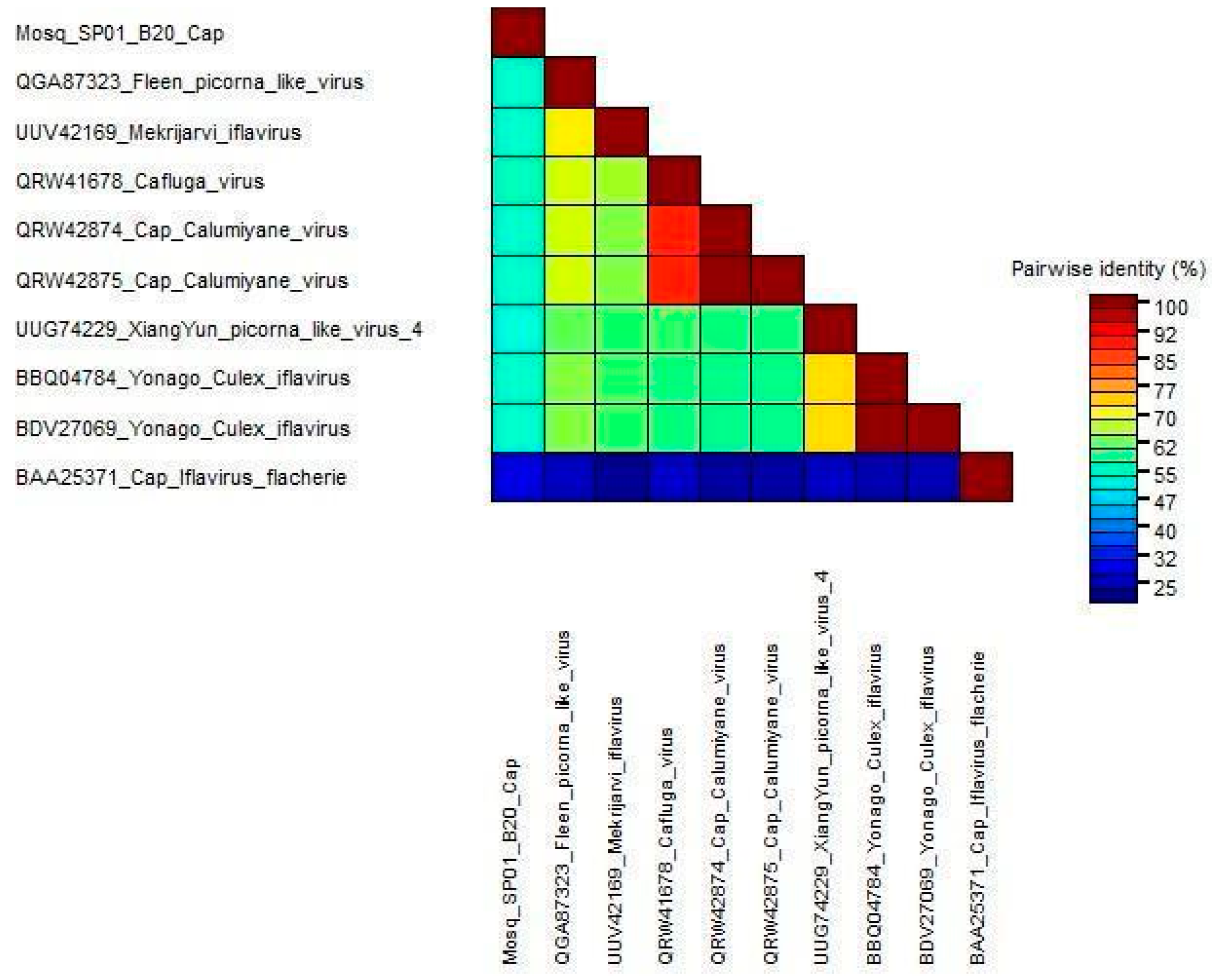
3.4. Genetic Diversity
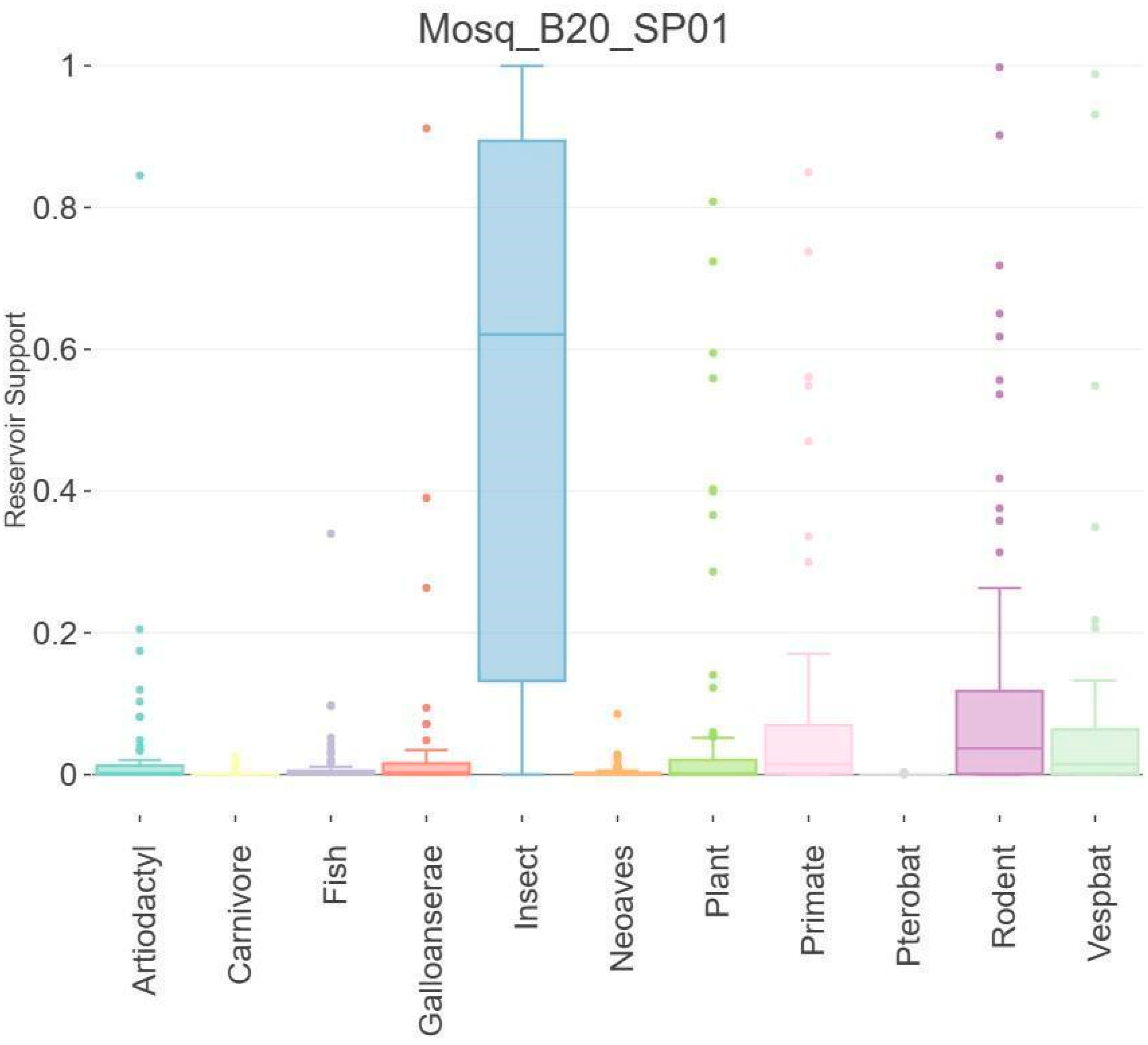
3.5. Host Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aragão, C.F.; Da Silva, S.P.; Do Nascimento, B.L.S.; Da Silva, F.S.; Nunes Neto, J.P.; Pinheiro, V.C.S.; Cruz, A.C.R. Shotgun Metagenomic Sequencing Reveals Virome Composition of Mosquitoes from a Transition Ecosystem of North-Northeast Brazil. Genes 2023, 14, 1443. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Lee, S.; Martins Lana, R.; Torres Codeço, C.; Castro, M.C.; Pascual, M. Emerging Arboviruses in the Urbanized Amazon Rainforest. BMJ 2020, 371, m4385. [Google Scholar] [CrossRef] [PubMed]
- Batson, J.; Dudas, G.; Haas-Stapleton, E.; Kistler, A.L.; Li, L.M.; Logan, P.; Ratnasiri, K.; Retallack, H. Single Mosquito Metatranscriptomics Identifies Vectors, Emerging Pathogens and Reservoirs in One Assay. eLife 2021, 10, e68353. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Yin, Q.; Zhou, L.; Meng, L.; Hu, W.; Li, F.; Li, Y.; Han, K.; Zhang, S.; Fu, S.; et al. Metagenomic Sequencing Reveals Viral Abundance and Diversity in Mosquitoes from the Shaanxi-Gansu-Ningxia Region, China. PLoS Negl. Trop. Dis. 2021, 15, e0009381. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the Invertebrate RNA Virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Sadeghi, M.; Altan, E.; Deng, X.; Barker, C.M.; Fang, Y.; Coffey, L.L.; Delwart, E. Virome of >12 Thousand Culex Mosquitoes from throughout California. Virology 2018, 523, 74–88. [Google Scholar] [CrossRef]
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented Genomic Diversity of RNA Viruses in Arthropods Reveals the Ancestry of Negative-Sense RNA Viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Frey, K.G.; Biser, T.; Hamilton, T.; Santos, C.J.; Pimentel, G.; Mokashi, V.P.; Bishop-Lilly, K.A. Bioinformatic Characterization of Mosquito Viromes within the Eastern United States and Puerto Rico: Discovery of Novel Viruses. Evol. Bioinform. 2016, 12s2, EBO.S38518. [Google Scholar] [CrossRef]
- Junglen, S.; Drosten, C. Virus Discovery and Recent Insights into Virus Diversity in Arthropods. Curr. Opin. Microbiol. 2013, 16, 507–513. [Google Scholar] [CrossRef]
- Webster, C.L.; Longdon, B.; Lewis, S.H.; Obbard, D.J. Twenty-Five New Viruses Associated with the Drosophilidae (Diptera). Evol. Bioinform. 2016, 12s2, EBO.S39454. [Google Scholar] [CrossRef]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; De Miranda, J.R.; Ryabov, E. ICTV Report Consortium ICTV Virus Taxonomy Profile: Iflaviridae. J. Gen. Virol. 2017, 98, 527–528. [Google Scholar] [CrossRef]
- Van Oers, M.M. Chapter in Book: Genomics and Biology of Iflaviruses, 231–250, Insect Virology; Asgari, S., Johnson, K.N., Eds.; Caister Academic: Norfolk, UK, 2010; ISBN 9781904455714. [Google Scholar]
- Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. A Conserved NTP-Motif in Putative Helicases. Nature 1988, 333, 22. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Xia, H.; Dong, C.; Cheng, Z.; Xia, X.; Zhang, J.; Zhou, X.; Hu, Y. Identification and Characterization of Iflavirus 3C-like Protease Processing Activities. Virology 2012, 428, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Morris, T.J. Evolution and Taxonomy of Positive-Strand RNA Viruses: Implications of Comparative Analysis of Amino Acid Sequences. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 375–430. [Google Scholar] [CrossRef]
- Ongus, J.R.; Roode, E.C.; Pleij, C.W.A.; Vlak, J.M.; Van Oers, M.M. The 5′ Non-Translated Region of Varroa Destructor Virus 1 (Genus Iflavirus): Structure Prediction and IRES Activity in Lymantria Dispar Cells. J. Gen. Virol. 2006, 87, 3397–3407. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Hu, Y.; Hu, L.; Zong, S.; Cai, D.; Wang, J.; Yu, H.; Zhang, J. Ectropis Obliqua Picorna-like Virus IRES-Driven Internal Initiation of Translation in Cell Systems Derived from Different Origins. J. Gen. Virol. 2007, 88, 2834–2838. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.R.; Dainat, B.; Locke, B.; Cordoni, G.; Berthoud, H.; Gauthier, L.; Neumann, P.; Budge, G.E.; Ball, B.V.; Stoltz, D.B. Genetic Characterization of Slow Bee Paralysis Virus of the Honeybee (Apis mellifera L.). J. Gen. Virol. 2010, 91, 2524–2530. [Google Scholar] [CrossRef]
- Isawa, H.; Asano, S.; Sahara, K.; Iizuka, T.; Bando, H. Analysis of Genetic Information of an Insect Picorna-like Virus, Infectious Flacherie Virus of Silkworm: Evidence for Evolutionary Relationships among Insect, Mammalian and Plant Picorna(-like) Viruses. Arch. Virol. 1998, 143, 127–143. [Google Scholar] [CrossRef]
- Patterson, E.I.; Kautz, T.F.; Contreras-Gutierrez, M.A.; Guzman, H.; Tesh, R.B.; Hughes, G.L.; Forrester, N.L. Negeviruses Reduce Replication of Alphaviruses during Coinfection. J. Virol. 2021, 95, e00433-21. [Google Scholar] [CrossRef]
- Baidaliuk, A.; Miot, E.F.; Lequime, S.; Moltini-Conclois, I.; Delaigue, F.; Dabo, S.; Dickson, L.B.; Aubry, F.; Merkling, S.H.; Cao-Lormeau, V.-M.; et al. Cell-Fusing Agent Virus Reduces Arbovirus Dissemination in Aedes aegypti Mosquitoes In Vivo. J. Virol. 2019, 93, e00705-19. [Google Scholar] [CrossRef]
- Romo, H.; Kenney, J.L.; Blitvich, B.J.; Brault, A.C. Restriction of Zika Virus Infection and Transmission in Aedes aegypti Mediated by an Insect-Specific Flavivirus. Emerg. Microbes Infect. 2018, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ramos, B.A.; Chagas, L.L.D.; De Arruda E Silva, F.; Santos, E.B.D.; Chiang, J.O.; Neto, J.P.N.; Vieira, D.B.R.; Junior, J.W.R.; Da Silva, E.V.P.; Freitas, M.N.O.; et al. Arboviruses in Free-Ranging Birds and Hematophagous Arthropods (Diptera, Nematocera) from Forest Remnants and Urbanized Areas of an Environmental Protection Area in the Amazon Biome. Viruses 2022, 14, 2101. [Google Scholar] [CrossRef]
- Araújo, P.A.; Freitas, M.O.; Chiang, J.O.; Silva, F.A.; Chagas, L.L.; Casseb, S.M.; Silva, S.P.; Nunes-Neto, J.P.; Rosa-Júnior, J.W.; Nascimento, B.S.; et al. Investigation about the Occurrence of Transmission Cycles of Arbovirus in the Tropical Forest, Amazon Region. Viruses 2019, 11, 774. [Google Scholar] [CrossRef]
- Silva, F.A.; Ferreira, M.S.; Araújo, P.A.; Casseb, S.M.M.; Silva, S.P.; Nunes Neto, J.P.; Chiang, J.O.; Rosa Junior, J.W.; Chagas, L.L.; Freitas, M.N.O.; et al. Serological and Molecular Evidence of the Circulation of the Venezuelan Equine Encephalitis Virus Subtype IIIA in Humans, Wild Vertebrates and Mosquitos in the Brazilian Amazon. Viruses 2022, 14, 2391. [Google Scholar] [CrossRef]
- Neto, J.P.N.; Reis, L.A.M.; Freitas, M.N.O.; Do Nascimento, B.L.S.; Das Chagas, L.L.; Da Costa, H.H.M.; Rodrigues, J.C.P.; Braga, C.M.; Da Silva, E.V.P.; Silva, S.P.; et al. First Isolation and Genome Sequence Analysis of West Nile Virus in Mosquitoes in Brazil. Trop. Med. Infect. Dis. 2023, 8, 237. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, L.D.O.; Simões, R.F.; Chagas, C.R.F.; Menezes, R.M.T.D.; Silva, F.S.; Monteiro, E.F.; Holcman, M.M.; Bajay, M.M.; Pinter, A.; Camargo-Neves, V.L.F.D.; et al. Assessing Diversity, Plasmodium Infection and Blood Meal Sources in Mosquitoes (Diptera: Culicidae) from a Brazilian Zoological Park with Avian Malaria Transmission. Insects 2021, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Forattini, O.P. Culicidologia Médica; Edusp: São Paulo, Brazil, 2002; ISBN 9788531406997. [Google Scholar]
- Consoli, R.A.G.B.; Oliveira, R.L.d. Principais Mosquitos de Importância Sanitária No Brasil; Editora Fiocruz: Rio de Janeiro, Brazil, 1994; ISBN 9788585676032. [Google Scholar]
- Lane, J. Neotropical Culicidae. São Paulo, v.2 p Ilus; Universidade de São Paulo—USP: São Paulo, Brazil, 1953; Volume 2. [Google Scholar]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rnaSPAdes: A de Novo Transcriptome Assembler and Its Application to RNA-Seq Data. GigaScience 2019, 8, giz100. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A Fast and Scalable Metagenome Assembler Driven by Advanced Methodologies and Community Practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Myung, I.J. Tutorial on Maximum Likelihood Estimation. J. Math. Psychol. 2003, 47, 90–100. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783. [Google Scholar] [CrossRef]
- Rombel, I.T.; Sykes, K.F.; Rayner, S.; Johnston, S.A. ORF-FINDER: A Vector for High-Throughput Gene Identification. Gene 2002, 282, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The Conserved Domain Database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The Conserved Domain Database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional Classification of Proteins via Subfamily Domain Architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Babayan, S.A.; Orton, R.J.; Streicker, D.G. Predicting Reservoir Hosts and Arthropod Vectors from Evolutionary Signatures in RNA Virus Genomes. Science 2018, 362, 577–580. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guimarães, L.d.O.; Bahia, S.L.; Ribeiro, G.d.O.; Ramos, E.d.S.F.; Villanova, F.; dos Santos Morais, V.; Telles-de-Deus, J.; Helfstein, V.C.; Santos, J.M.d.; Pandey, R.P.; et al. New Iflavirus Species Characterized from Mosquitoes Captured in the Sao Paulo Zoological Facilities. Microorganisms 2024, 12, 1749. https://doi.org/10.3390/microorganisms12091749
Guimarães LdO, Bahia SL, Ribeiro GdO, Ramos EdSF, Villanova F, dos Santos Morais V, Telles-de-Deus J, Helfstein VC, Santos JMd, Pandey RP, et al. New Iflavirus Species Characterized from Mosquitoes Captured in the Sao Paulo Zoological Facilities. Microorganisms. 2024; 12(9):1749. https://doi.org/10.3390/microorganisms12091749
Chicago/Turabian StyleGuimarães, Lilian de Oliveira, Santana Lobato Bahia, Geovani de Oliveira Ribeiro, Endrya do Socorro Foro Ramos, Fabiola Villanova, Vanessa dos Santos Morais, Juliana Telles-de-Deus, Vanessa Christe Helfstein, Jesus Maia dos Santos, Ramendra Pati Pandey, and et al. 2024. "New Iflavirus Species Characterized from Mosquitoes Captured in the Sao Paulo Zoological Facilities" Microorganisms 12, no. 9: 1749. https://doi.org/10.3390/microorganisms12091749