
The Response of Rhizosphere Microbial C and N-Cycling Gene Abundance of Sand-Fixing Shrub to Stand Age Following Desert Restoration

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Area
2.2. Site Selection and Soil Sampling Preparation
2.3. Determination of Soil Physicochemical Properties
2.4. Shotgun Metagenome Sequencing and Sequence Processing
2.5. Carbon and Nitrogen Functional Gene Screening from Shotgun Metagenome Sequence
2.6. Statistical Analysis
3. Results
3.1. Characteristics of Soil Abiotic and Biotic Factors
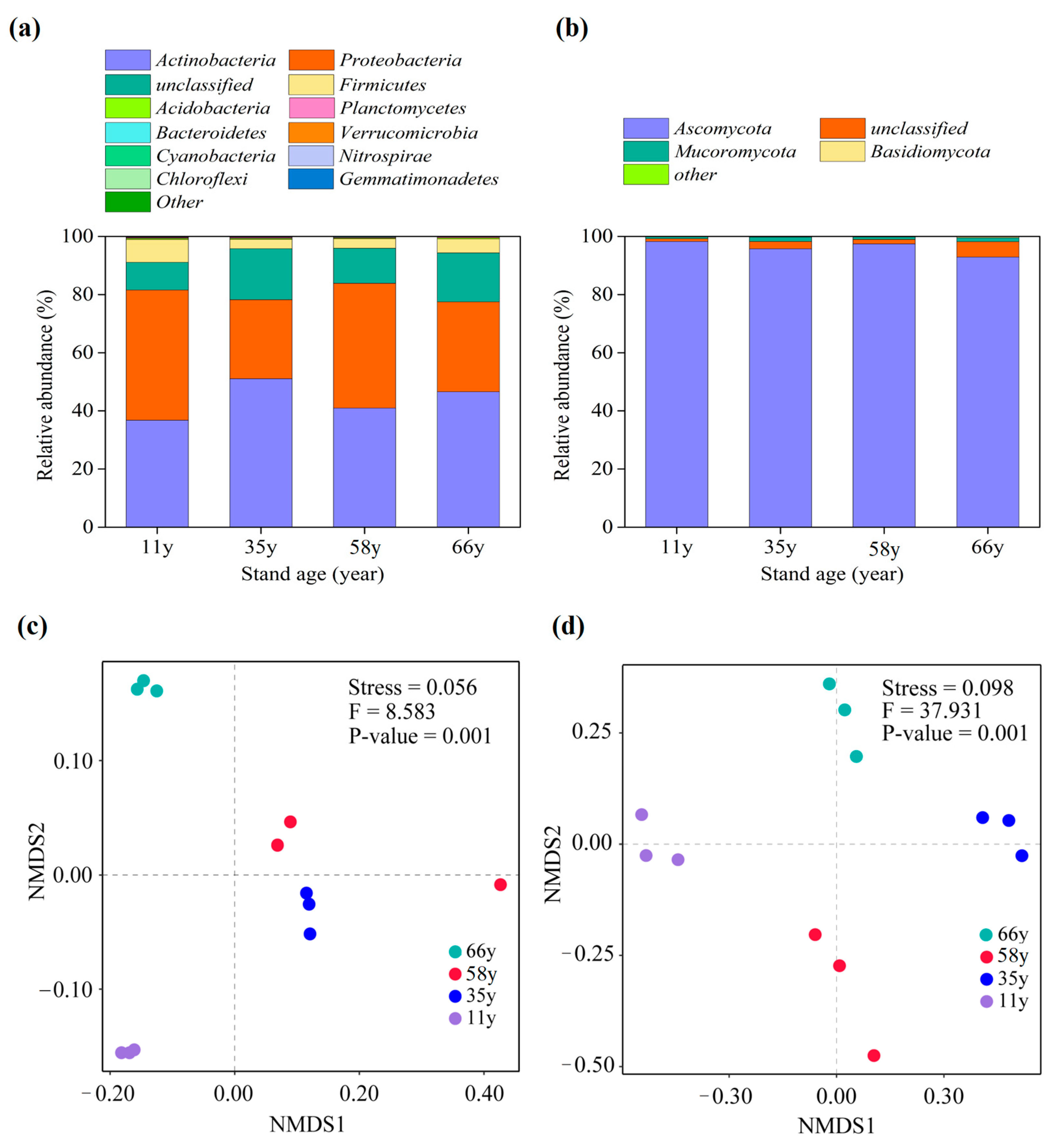
3.2. Soil Microbial Community Composition Changes along Stand Age
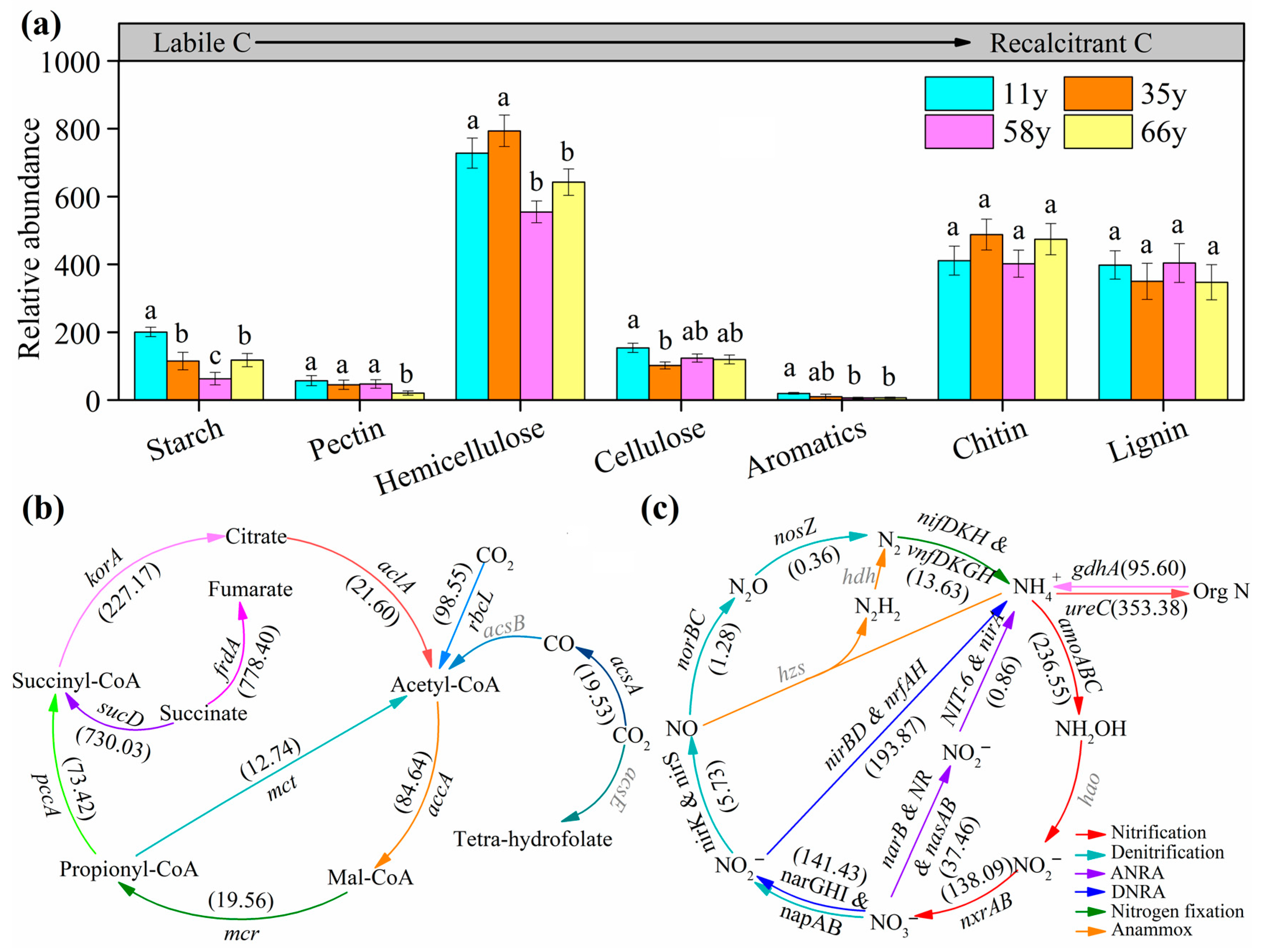
3.3. Soil Microbial Functional Gene Abundance along Stand Age
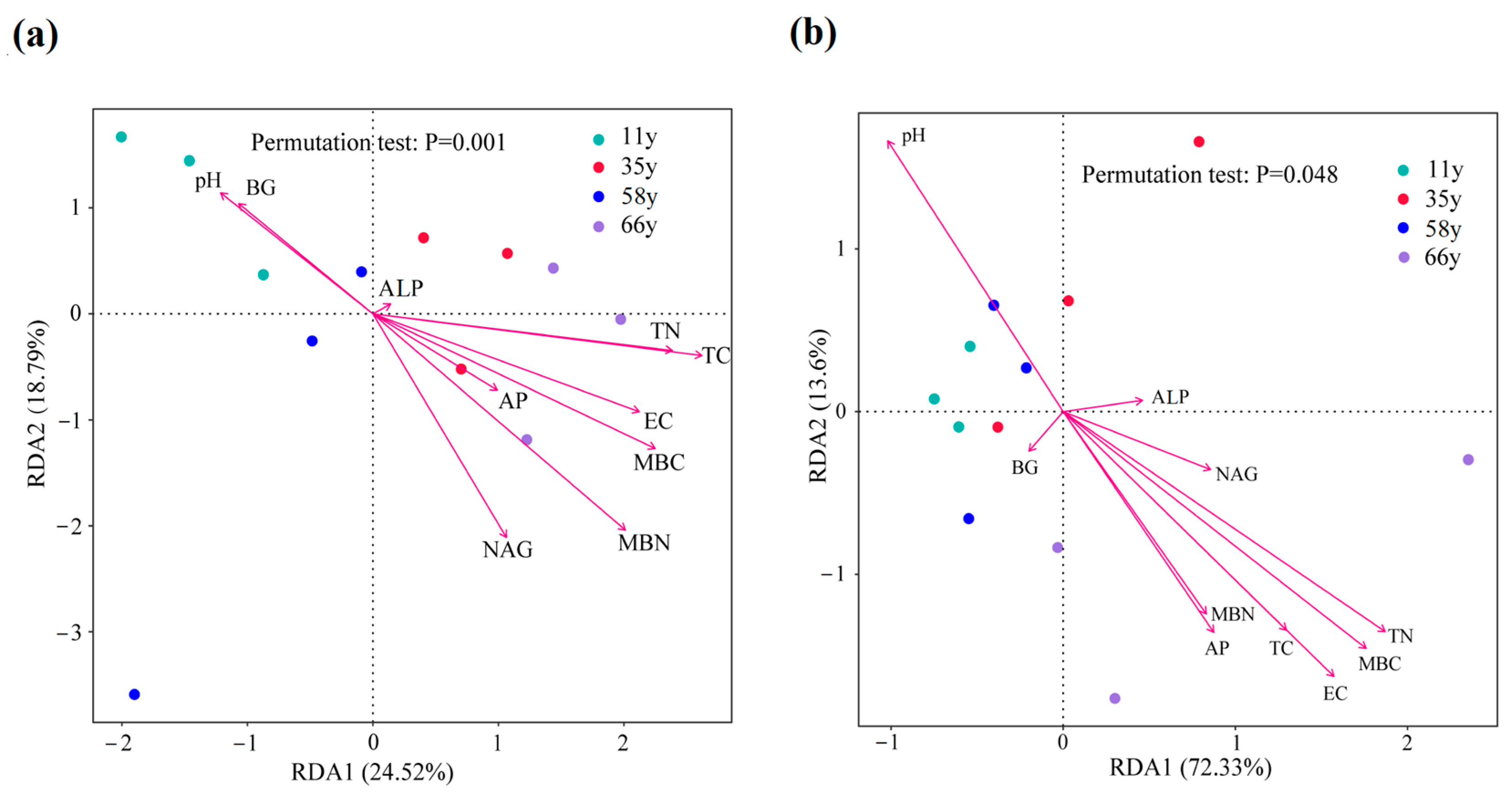
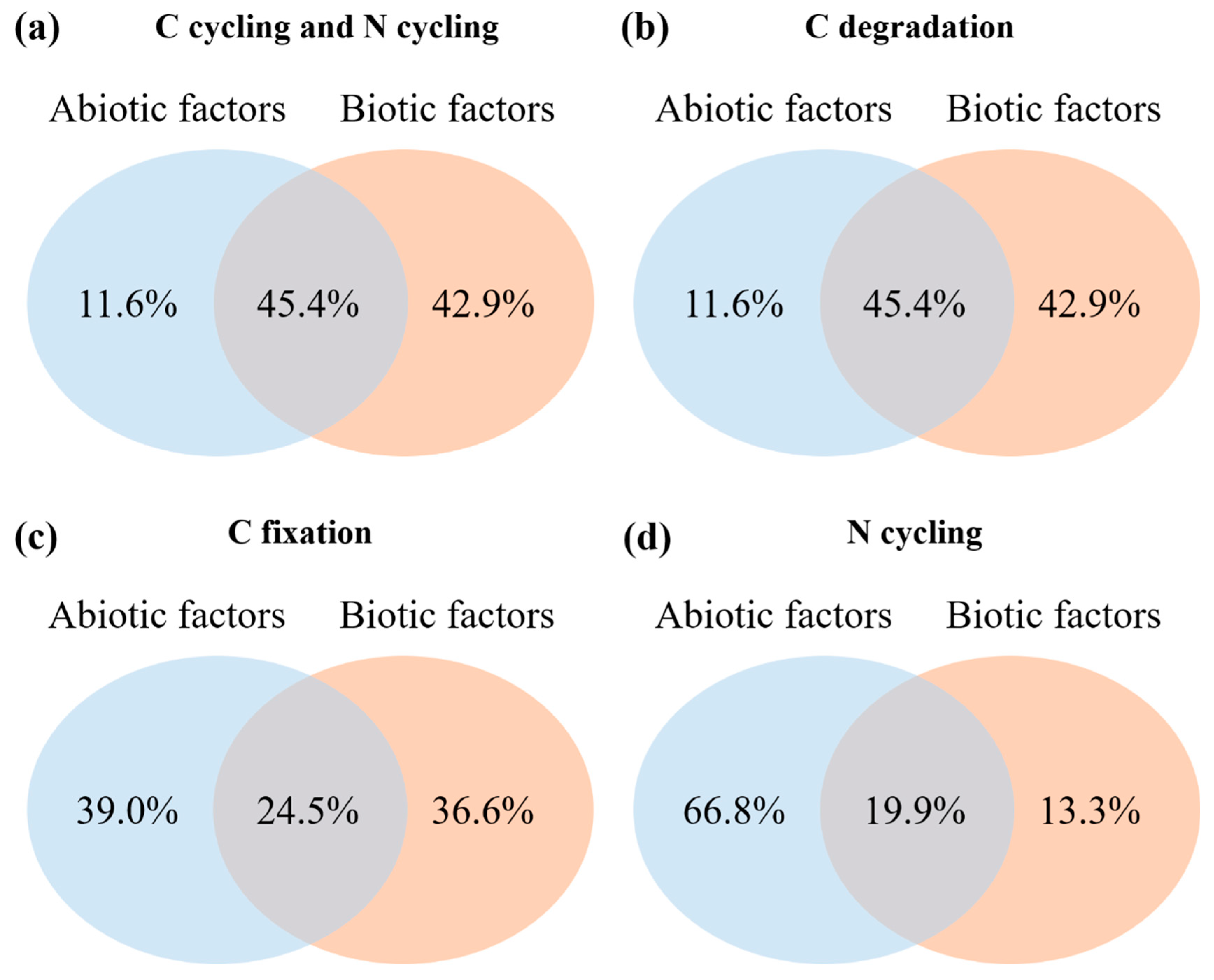
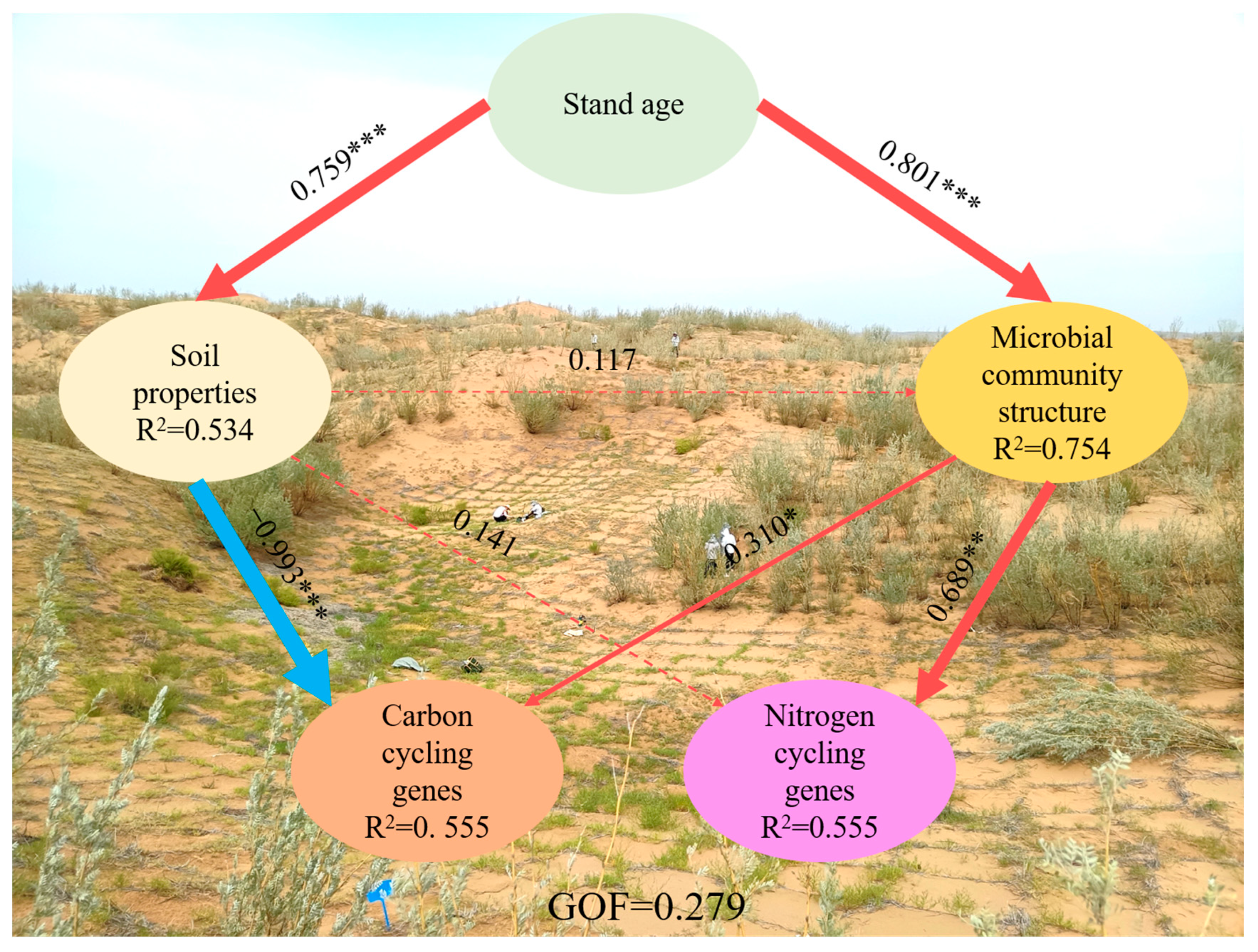
3.4. Impacts of Environmental Variables on Rhizosphere Soil Microbial Community Composition and Functional Gene Abundance
4. Discussion
4.1. Effects of Stand Age on Soil Properties
4.2. Effects of Stand Age on the Soil Microbial Community and the Genes Related to C and N Cycling
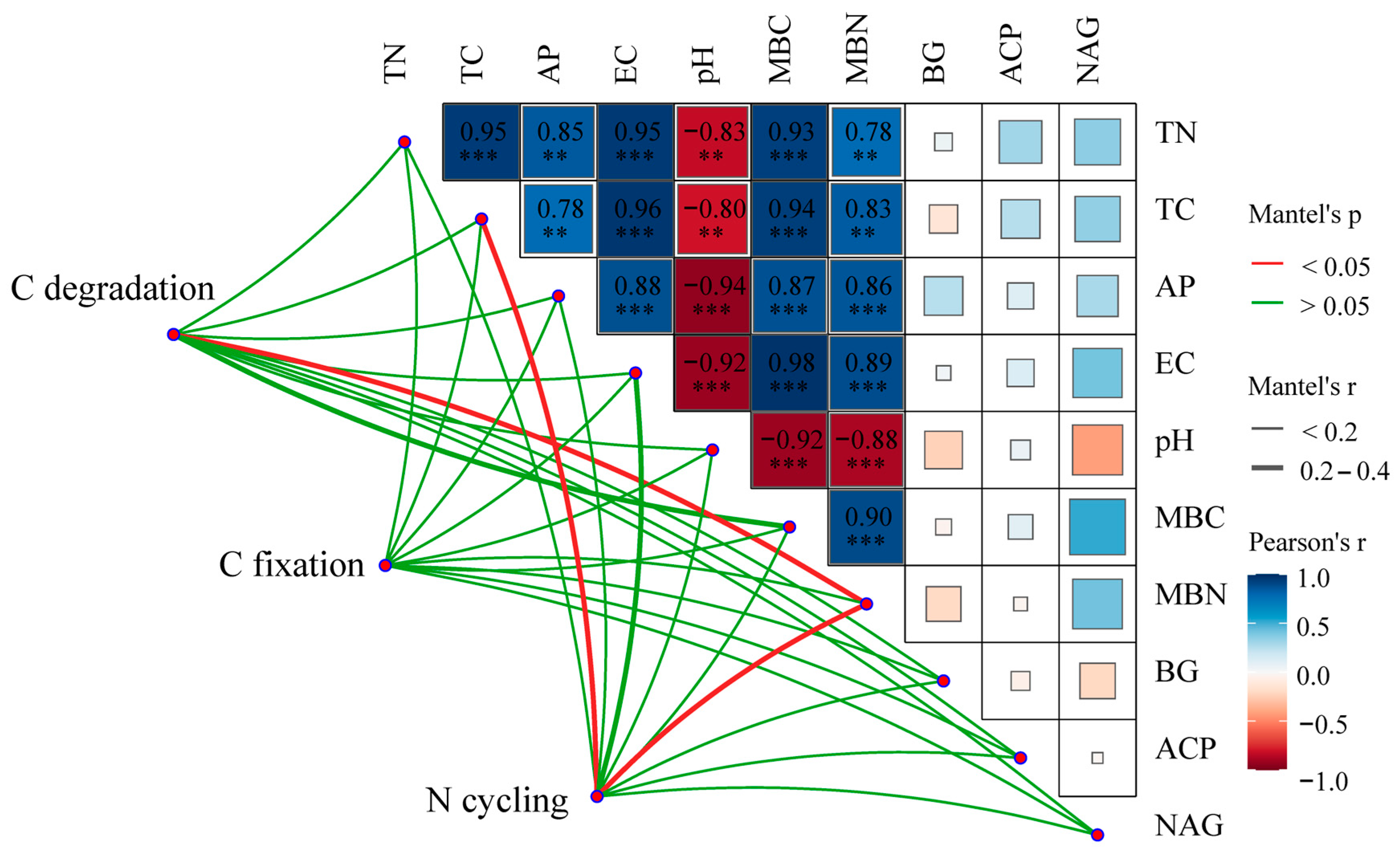
4.3. Linkages between Rhizosphere Soil Microbial and Environmental Factors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Shao, P.; Liang, C.; Lynch, L.; Xie, H.; Bao, X. Reforestation accelerates soil organic carbon accumulation: Evidence from microbial biomarkers. Soil Biol. Biochem. 2019, 131, 182–190. [Google Scholar] [CrossRef]
- Lu, Z.X.; Wang, P.; Ou, H.B.; Wei, S.X.; Wu, L.C.; Jiang, Y.; Wang, R.J.; Liu, X.S.; Wang, Z.H.; Chen, L.J.; et al. Effects of different vegetation restoration on soil nutrients, enzyme activities, and microbial communities in degraded karst landscapes in southwest China. For. Ecol. Manag. 2022, 508, 120002. [Google Scholar] [CrossRef]
- Li, S.; Liber, K. Influence of different revegetation choices on plant community and soil development nine years after initial planting on a reclaimed coal gob pile in the Shanxi mining area, China. Sci. Total Environ. 2018, 618, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Y.; Jia, X.; An, Z. Impacts of shrub introduction on soil properties and implications for dryland revegetation. Sci. Total Environ. 2020, 742, 140498. [Google Scholar] [CrossRef]
- Du, L.; Zeng, Y.; Ma, L.; Qiao, C.; Wu, H.; Su, Z.; Bao, G. Effects of anthropogenic revegetation on the water and carbon cycles of a desert steppe ecosystem. Agric. For. Meteorol. 2021, 300, 108339. [Google Scholar] [CrossRef]
- Li, J.; Xie, T.; Zhu, H.; Zhou, J.; Li, C.; Xiong, W.; Xu, L.; Wu, Y.; He, Z.; Li, X. Alkaline phosphatase activity mediates soil organic phosphorus mineralization in a subalpine forest ecosystem. Geoderma 2021, 404, 115376. [Google Scholar] [CrossRef]
- Pietri, J.A.; Brookes, P.C. Substrate inputs and pH as factors controlling microbial biomass, activity and community structure in an arable soil. Soil Biol. Biochem. 2009, 41, 1396–1405. [Google Scholar] [CrossRef]
- Xu, Z.; Yu, G.; Zhang, X.; Ge, J.; He, N.; Wang, Q.; Wang, D. The variations in soil microbial communities, enzyme activities and their relationships with soil organic matter decomposition along the northern slope of Changbai Mountain. Appl. Soil Ecol. 2015, 86, 19–29. [Google Scholar] [CrossRef]
- Luo, Z.; Liu, J.; Jia, T.; Chai, B.; Wu, T. Soil bacterial community response and nitrogen cycling variations associated with subalpine meadow degradation on the Loess Plateau, China. Appl. Environ. Microbiol. 2020, 86, e00180-20. [Google Scholar] [CrossRef]
- Liao, J.; Dou, Y.; Yang, X.; An, S. Soil microbial community and their functional genes during grassland restoration. J. Environ. Manag. 2023, 325, 116488. [Google Scholar] [CrossRef]
- Fuchslueger, L.; Wild, B.; Mooshammer, M.; Takriti, M.; Kienzl, S.; Knoltsch, A.; Hofhansla, F.; Bahn, M.; Richter, A. Microbial carbon and nitrogen cycling responses to drought and temperature in differently managed mountain grasslands. Soil Biol. Biochem. 2019, 135, 144–153. [Google Scholar] [CrossRef]
- Maestre, F.T.; Delgado-Baquerizo, M.; Jeffries, T.C.; Eldridge, D.J.; Ochoa, V.; Gozalo, B.; Quero, J.L.; García-Gómez, M.; Gallardo, A.; Ulrich, W.; et al. Increasing aridity reduces soil microbial diversity and abundance in global drylands. Proc. Natl. Acad. Sci. USA 2015, 112, 15684–15689. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Wang, F.; Hu, C.; Liu, B. Metagenomics reveals taxon-specific responses of the nitrogen-cycling microbial community to long-term nitrogen fertilization. Soil Biol. Biochem. 2021, 156, 108214. [Google Scholar] [CrossRef]
- Chen, J.; Sinsabaugh, R.L. Linking microbial functional gene abundance and soil extracellular enzyme activity: Implications for soil carbon dynamics. Glob. Change Biol. 2021, 27, 1322–1325. [Google Scholar] [CrossRef]
- Li, W.; Li, Y.; Lv, J.; He, X.; Wang, J.; Teng, D.; Jiang, L.; Wang, H.; Lv, G. Rhizosphere effect alters the soil microbiome composition and C, N transformation in an arid ecosystem. Appl. Soil Ecol. 2022, 170, 104296. [Google Scholar] [CrossRef]
- Vries, F.T.; Griffths, R.I.; Knight, C.G.; Nicolitch, O.; Williams, A. Harnessing rhizosphere microbiomes for drought-resilient crop production. Science 2020, 368, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Beckers, B.; Op De Beeck, M.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Huang, X.; Zhang, J.; Cai, Z.; Jiang, K.; Chang, Y. Deciphering the relative importance of soil and plant traits on the development of rhizosphere microbial communities. Soil Biol. Biochem. 2020, 148, 107909. [Google Scholar] [CrossRef]
- Raaijmakers, J.M.; Mazzola, M. Soil immune responses. Science 2016, 352, 1392–1393. [Google Scholar] [CrossRef]
- Zhalnina, K.; Louie, K.B.; Hao, Z.; Mansoori, N.; Da Rocha, U.N.; Shi, S.; Cho, H.; Karaoz, U.; Loqué, D.; Bowen, B.P.; et al. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 2018, 3, 470–480. [Google Scholar] [CrossRef]
- Schmidt, J.E.; Kent, A.D.; Brisson, V.L.; Gaudin, A. Agricultural management and plant selection interactively affect rhizosphere microbial community structure and nitrogen cycling. Microbiome 2019, 7, 146. [Google Scholar] [CrossRef]
- Kuzyakov, Y.; Blagodatskaya, E. Microbial hotspots and hot moments in soil: Concept & review. Soil Biol. Biochem. 2015, 83, 184–199. [Google Scholar] [CrossRef]
- Zhang, X.; Dippold, M.A.; Kuzyakov, Y.; Razavi, B.S. Spatial pattern of enzyme activities depends on root exudate composition. Soil Biol. Biochem. 2019, 133, 83–93. [Google Scholar] [CrossRef]
- Yang, H.; Li, X.; Wang, Z.; Jia, R.; Liu, L.; Chen, Y.; Wei, Y.; Gao, Y.; Li, G. Carbon sequestration capacity of shifting sand dune after establishing new vegetation in the Tengger Desert, northern China. Sci. Total Environ. 2014, 478, 1–11. [Google Scholar] [CrossRef]
- Liu, J.; Qiu, L.; Wang, X.; Wei, X.; Gao, H.; Zhang, Y.; Cheng, J. Effects of wildfire and topography on soil nutrients in a semiarid restored grassland. Plant Soil 2018, 428, 123–136. [Google Scholar] [CrossRef]
- Chen, W.; Yu, T.; Zhao, C.; Li, B.; Qin, Y.; Li, H.; Tang, H.; Liu, J.; Zhang, X. Development and determinants of topsoil bacterial and fungal communities of afforestation by aerial sowing in Tengger Desert, China. J. Fungi 2023, 9, 399. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, Y.; Wang, Z.; Yuan, S.; Qi, J.; Zhang, W.; Wang, Y.; Li, X. Bacteria and fungi differentially contribute to carbon and nitrogen cycles during biological soil crust succession in arid ecosystems. Plant Soil 2020, 447, 379–392. [Google Scholar] [CrossRef]
- Li, X.R.; Zhang, Z.S.; Liu, Y.B.; Li, X.J.; Yang, H.T. Fundamental Ecohydrology of Ecological Restoration and Recovery in Sandy Desert Regions of China; Science Press in Chinese: Beijing, China, 2016. [Google Scholar]
- Schmidt, C.S.; Richardson, D.J.; Baggs, E.M. Constraining the conditions conducive to dissimilatory nitrate reduction to ammonium in temperate arable soils. Soil Biol. Biochem. 2011, 43, 1607–1611. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Xu, X.L.; Wang, H.M.; Blagodatskaya, E.; Kuzyakov, Y. Dominant extracellular enzymes in priming of SOM decomposition depend on temperature. Geoderma 2019, 343, 187–195. [Google Scholar] [CrossRef]
- Yi, S.; Jin, W.T.; Yuan, Y.N.; Fang, Y.H. An optimized CTAB method for genomic DNA extraction from freshly-picked pinnae of fern. Adiantum capillus-veneris L. BioProtocol 2018, 8, 2906. [Google Scholar] [CrossRef]
- Clark, M.J.; Chen, R.; Lam, H.Y.K.; Karczewski, K.J.; Chen, R.; Euskirchen, G.; Butte, A.J.; Snyder, M. Performance comparison of exome DNA sequencing technologies. Nat. biotechnol. 2011, 29, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Ramayah, T.; Cheah, J.; Chuah, F.; Ting, H.; Memon, M.A. Partial Least Squares Structural Equation Modeling (PLS-SEM) Using SmartPLS 3.0: An Updated Guide and Practical Guide to Statistical Analysis, 2nd ed.; Pearson: Kuala Lumpur, Malaysia, 2018. [Google Scholar]
- Panchal, P.; Preece, C.; Peñuelas, J.; Giri, J. Soil carbon sequestration by root exudates. Trends Plant Sci. 2022, 27, 749–757. [Google Scholar] [CrossRef]
- Raza, A.; Zahra, N.; Hafeez, M.B.; Ahmad, M.; Iqbal, S.; Shaukat, K.; Ahmad, G. Nitrogen fixation of legumes: Biology and Physiology. In The Plant Family Fabaceae: Biology and Physiological Responses to Environmental Stresses; Springer: Singapore, 2020; pp. 43–74. [Google Scholar]
- Clarholm, M.; Skyllberg, U.; Rosling, A. Organic acid induced release of nutrients from metal-stabilized soil organic matter–the unbutton model. Soil Biol. Biochem. 2015, 84, 168–176. [Google Scholar] [CrossRef]
- Pantigoso, H.A.; Manter, D.K.; Fonte, S.J.; Vivanco, J.M. Root exudate-derived compounds stimulate the phosphorus solubilizing ability of bacteria. Sci. Rep. 2023, 13, 4050. [Google Scholar] [CrossRef]
- Wen, T.; Yu, G.H.; Hong, W.D.; Yuan, J.; Niu, G.Q.; Xie, P.H.; Sun, F.S.; Guo, L.D.; Kuzyakov, Y.; Shen, Q.R. Root exudate chemistry affects soil carbon mobilization via microbial community reassembly. Fundam. Res. 2022, 2, 697–707. [Google Scholar] [CrossRef]
- Steinauer, K.; Chatzinotas, A.; Eisenhauer, N. Root exudate cocktails: The link between plant diversity and soil microorganisms? Ecol. Evol. 2016, 6, 7387–7396. [Google Scholar] [CrossRef]
- Girkin, N.T.; Turner, B.L.; Ostle, N.; Sjögersten, S. Composition and concentration of root exudate analogues regulate greenhouse gas fluxes from tropical peat. Soil Biol. Biochem. 2018, 127, 280–285. [Google Scholar] [CrossRef]
- Sasse, J.; Martinoia, E.; Northen, T. Feed your friends: Do plant exudates shape the root microbiome? Trends Plant Sci. 2018, 23, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Fang, L.; Yang, C.; Chen, W.; Cui, Y.; Li, S. Reveal the response of enzyme activities to heavy metals through in situ zymography. Ecotoxicol. Environ. Saf. 2018, 156, 106–115. [Google Scholar] [CrossRef]
- Rosinger, C.; Rousk, J.; Sandén, H. Can enzymatic stoichiometry be used to determine growth-limiting nutrients for microorganisms?-A critical assessment in two subtropical soils. Soil Biol. Biochem. 2019, 128, 115–126. [Google Scholar] [CrossRef]
- Liu, Y.; Evans, S.E.; Friesen, M.L.; Tiemann, L.K. Root exudates shift how N mineralization and N fixation contribute to the plant-available N supply in low fertility soils. Soil Biol. Biochem. 2022, 165, 108541. [Google Scholar] [CrossRef]
- Eo, J.; Park, K.C. Long-term effects of imbalanced fertilization on the composition and diversity of soil bacterial community. Agric. Ecosyst. Environ. 2016, 231, 176–182. [Google Scholar] [CrossRef]
- Tu, J.; Qiao, J.; Zhu, Z.; Li, P.; Wu, L. Soil bacterial community responses to long-term fertilizer treatments in Paulownia plantations in subtropical China. Appl. Soil Ecol. 2018, 124, 317–326. [Google Scholar] [CrossRef]
- Mohammadipanah, F.; Wink, J. Actinobacteria from arid and desert habitats: Diversity and biological activity. Front Microbiol. 2016, 6, 1541. [Google Scholar] [CrossRef]
- Francioli, D.; Rijssel, S.Q.; Ruijven, J.; Termorshuizen, A.J.; Cotton, T.A.; Dumbrell, A.J.; Raaijmakers, J.M.; Weigelt, A.; Mommer, L. Plant functional group drives the community structure of saprophytic fungi in a grassland biodiversity experiment. Plant Soil 2021, 461, 91–105. [Google Scholar] [CrossRef]
- Guo, X.; Feng, J.; Shi, Z.; Zhou, X.; Yuan, M.; Tao, X.; Hale, L.; Yuan, T.; Wang, J.; Qin, Y.; et al. Climate warming leads to divergent succession of grassland microbial communities. Nat. Clim. Change 2018, 8, 813–818. [Google Scholar] [CrossRef]
- Chanthorn, W.; Hartig, F.; Brockelman, W.Y. Structure and community composition in a tropical forest suggest a change of ecological processes during stand development. For. Ecol. Manag. 2017, 404, 100–107. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, H.; Zhao, P.; Wei, X.; Ding, G.; Gao, G.; Shi, M. Vegetation restoration alters fungal community composition and functional groups in a desert ecosystem. Front. Environ. Sci. 2021, 9, 589068. [Google Scholar] [CrossRef]
- Prado, I.G.O.; Silva, M.D.C.S.; Prado, D.G.O.; Kemmelmeier, K.; Pedrosa, B.G.; Silva, C.C.; Kasuya, M.C.M. Revegetation process increases the diversity of total and arbuscular mycorrhizal fungi in areas affected by the Fundão dam failure in Mariana, Brazil. Appl. Soil Ecol. 2019, 141, 84–95. [Google Scholar] [CrossRef]
- Crits-Christoph, A.; Olm, M.R.; Diamond, S.; Bouma-Gregson, K.; Banfield, J.F. Soil bacterial populations are shaped by recombination and gene-specific selection across a grassland meadow. ISME J. 2020, 14, 1834–1846. [Google Scholar] [CrossRef]
- Domeignoz-Horta, L.A.; Pold, G.; Liu, X.J.A.; Frey, S.D.; Melillo, J.M.; DeAngelis, K.M. Microbial diversity drives carbon use efficiency in a model soil. Nat. Commun. 2020, 11, 3684. [Google Scholar] [CrossRef]
- Li, J.; Fan, M.; Yang, L.; Yang, Z.; Shangguan, Z. Temporal shifts in root exudates driven by vegetation restoration alter rhizosphere microbiota in Robinia pseudoacacia plantations. Tree Physiol. 2023, 43, 1081–1091. [Google Scholar] [CrossRef]
- Treseder, K.K.; Berlemont, R.; Allison, S.D.; Martiny, A.C. Nitrogen enrichment shifts functional genes related to nitrogen and carbon acquisition in the fungal community. Soil Biol. Biochem. 2018, 123, 87–96. [Google Scholar] [CrossRef]
- Li, Z.; Zeng, Z.; Song, Z.; Wang, F.; Tian, D.; Mi, W.; Huang, X.; Wang, J.; Song, L.; Yang, Z.; et al. Vital roles of soil microbes in driving terrestrial nitrogen immobilization. Glob. Change Biol. 2021, 27, 1848–1858. [Google Scholar] [CrossRef]
- An, F.; Niu, Z.; Liu, T.; Su, Y. Succession of soil bacterial community along a 46-year choronsequence artificial revegetation in an arid oasis-desert ecotone. Sci. Total Environ. 2022, 814, 152496. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Liu, G.; Zhang, C. Response of rhizosphere microbial communities to plant succession along a grassland chronosequence in a semiarid area. J. Soils Sediments 2019, 19, 2496–2508. [Google Scholar] [CrossRef]
- Schlatter, D.C.; Kahl, K.; Carlson, B.; Huggins, D.R.; Paulitz, T. Fungal community composition and diversity vary with soil depth and landscape position in a no-till wheat-based cropping system. FEMS Microbiol. Ecol. 2018, 94, fiy098. [Google Scholar] [CrossRef]
- Zheng, Y.; Hu, H.W.; Guo, L.D.; Anderson, I.C.; Powell, J.R. Dryland forest management alters fungal community composition and decouples assembly of root-and soil-associated fungal communities. Soil Biol. Biochem. 2017, 109, 14–22. [Google Scholar] [CrossRef]
- Schappe, T.; Albornoz, F.E.; Turner, B.L.; Jones, F.A. Co-occurring fungal functional groups respond differently to tree neighborhoods and soil properties across three tropical rainforests in Panama. Microb. Ecol. 2020, 79, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Louca, S.; Doebeli, M. Taxonomic variability and functional stability in microbial communities infected by phages. Environ. Microbiol. 2017, 19, 3863–3878. [Google Scholar] [CrossRef] [PubMed]
- Starke, R.; Mondéjar, R.L.; Human, Z.R.; Navrátilová, D.; Štursová, M.; Větrovský, T.; Olson, H.M.; Orton, D.J.; Callister, S.J.; Lipton, M.S.; et al. Niche differentiation of bacteria and fungi in carbon and nitrogen cycling of different habitats in a temperate coniferous forest: A metaproteomic approach. Soil Biol. Biochem. 2021, 155, 108170. [Google Scholar] [CrossRef]
- Guo, X.; Zhou, X.; Hale, L.; Yuan, M.; Ning, D.; Feng, J.; Shi, Z.; Li, Z.; Feng, B.; Gao, Q.; et al. Climate warming accelerates temporal scaling of grassland soil microbial biodiversity. Nat. Ecol. Evol. 2019, 3, 612–619. [Google Scholar] [CrossRef]
- Yang, Y.; Dou, Y.; Wang, B.; Wang, Y.; Liang, C.; An, S.; Soromotin, A.; Kuzyakov, Y. Increasing contribution of microbial residues to soil organic carbon in grassland restoration chronosequence. Soil Biol. Biochem. 2022, 170, 108688. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Stand Age (Year) | ||||
|---|---|---|---|---|---|
| 11 y | 35 y | 58 y | 66 y | ||
| Abiotic factors | TN (g/kg) | 0.23 ± 0.01 b | 0.28 ± 0.03 b | 0.31 ± 0.05 b | 0.57 ± 0.03 a |
| TC (g/kg) | 3.86 ± 0.10 c | 4.60 ± 0.14 b | 4.56 ± 0.10 b | 7.08 ± 0.15 a | |
| C:N | 16.81 ± 0.70 a | 16.82 ± 1.00 a | 15.57 ± 2.38 a | 12.39 ± 0.63 a | |
| AP (mg/kg) | 72.21 ± 0.60 c | 70.64 ± 0.29 c | 78.92 ± 2.29 b | 85.53 ± 0.20 a | |
| EC (μS/cm) | 122.37 ± 2.38 c | 148.87 ± 5.63 c | 293.33 ± 2.19 b | 717.33 ± 27.53 a | |
| pH | 8.05 ± 0.02 b | 8.11 ± 0.01 a | 7.91 ± 0.01 c | 7.78 ± 0.02 d | |
| Biotic factors | MBC (mg/kg) | 49.03 ± 1.18 c | 65.28 ± 0.77 c | 97.80 ± 1.12 b | 164.38 ± 14.63 a |
| MBN (mg/kg) | 5.75 ± 0.48 d | 8.54 ± 0.69 c | 13.46 ± 0.69 b | 17.41 ± 0.73 a | |
| BG (mg/g/h) | 0.30 ± 0.01 a | 0.12 ± 0.02 c | 0.22 ± 0.04 b | 0.22 ± 0.01 b | |
| ALP (μg/g/h) | 35.03 ± 2.73 a | 37.38 ± 5.44 a | 31.10 ± 4.40 a | 37.46 ± 1.70 a | |
| NAG (μmol/d/g) | 5.35 ± 1.04 a | 6.09 ± 0.52 a | 6.62 ± 0.73 a | 7.18 ± 1.58 a | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wang, B.; Wang, Z.; He, W.; Wang, Y.; Liu, L.; Yang, H. The Response of Rhizosphere Microbial C and N-Cycling Gene Abundance of Sand-Fixing Shrub to Stand Age Following Desert Restoration. Microorganisms 2024, 12, 1752. https://doi.org/10.3390/microorganisms12091752
Li Y, Wang B, Wang Z, He W, Wang Y, Liu L, Yang H. The Response of Rhizosphere Microbial C and N-Cycling Gene Abundance of Sand-Fixing Shrub to Stand Age Following Desert Restoration. Microorganisms. 2024; 12(9):1752. https://doi.org/10.3390/microorganisms12091752
Chicago/Turabian StyleLi, Yunfei, Bingyao Wang, Zhanjun Wang, Wenqiang He, Yanli Wang, Lichao Liu, and Haotian Yang. 2024. "The Response of Rhizosphere Microbial C and N-Cycling Gene Abundance of Sand-Fixing Shrub to Stand Age Following Desert Restoration" Microorganisms 12, no. 9: 1752. https://doi.org/10.3390/microorganisms12091752