
Rhizosphere Microbiome Co-Occurrence Network Analysis across a Tomato Domestication Gradient


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant and Soil Selection
2.2. Tomato Growth Conditions
2.3. Collection and DNA Extraction of Rhizosphere Soil Samples
2.4. PCR and minION Sequencing Conditions
2.5. Co-Occurrence Network Generation
2.6. Data Analysis
3. Results
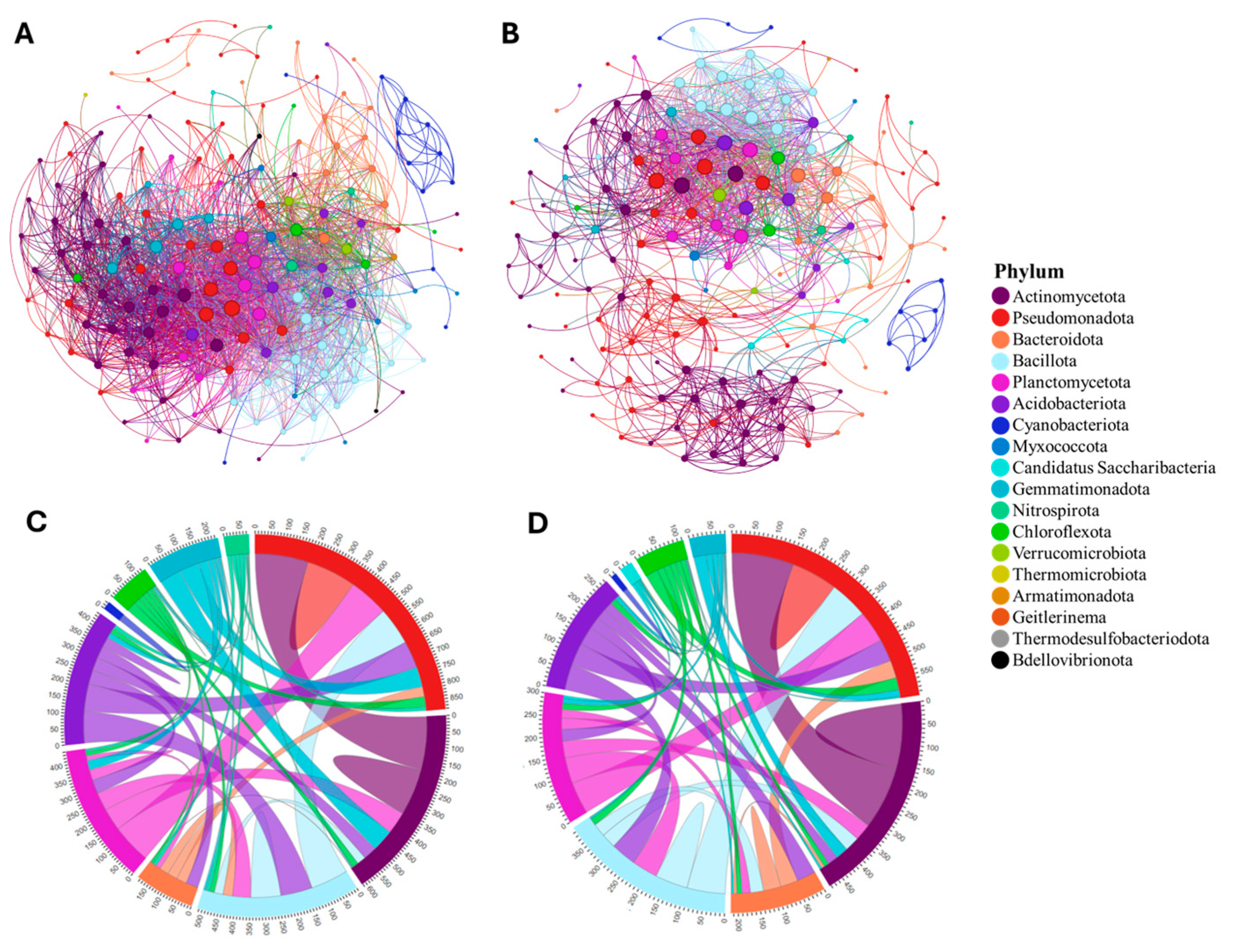
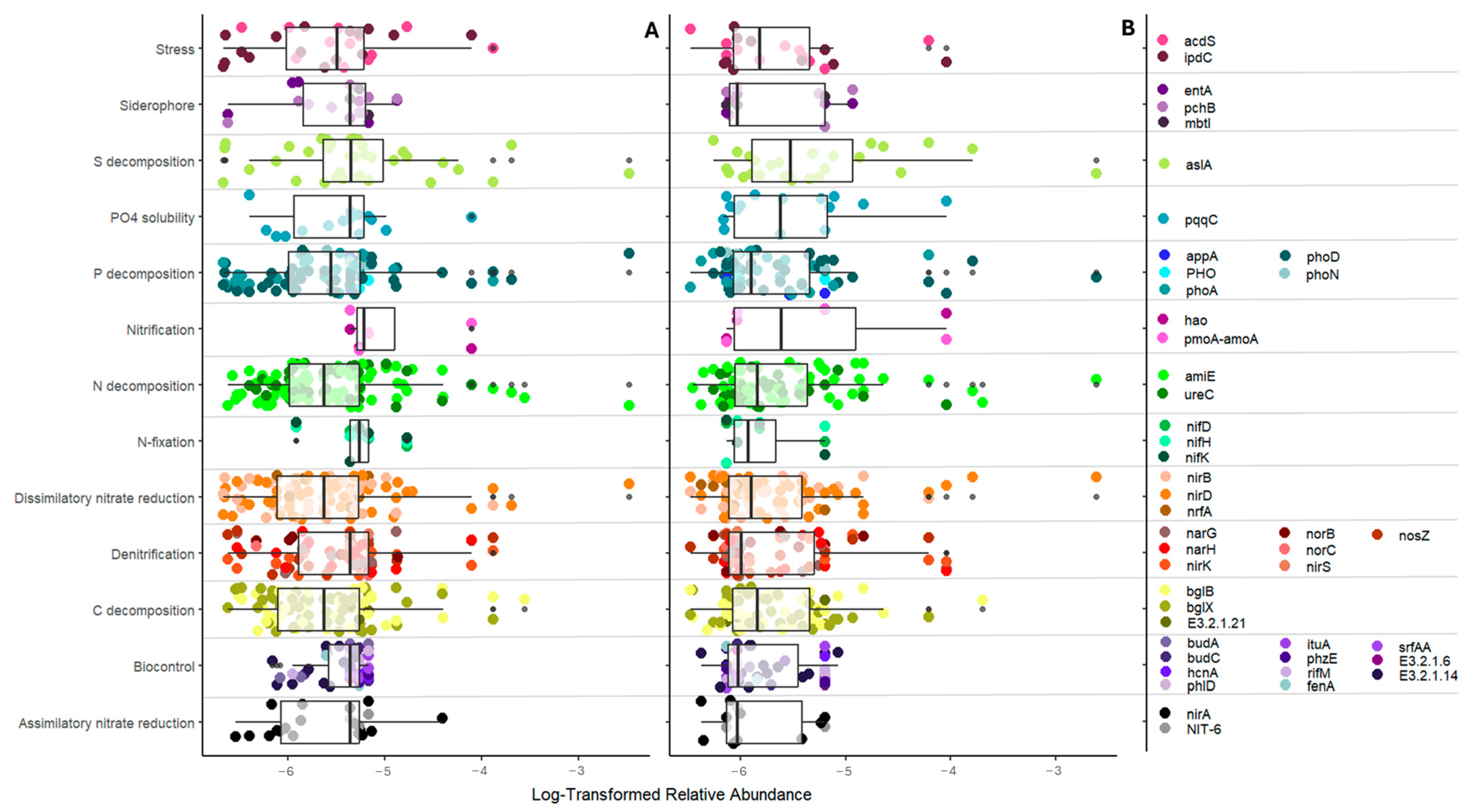
3.1. Soil Bacterial Community Interactions and Functionality
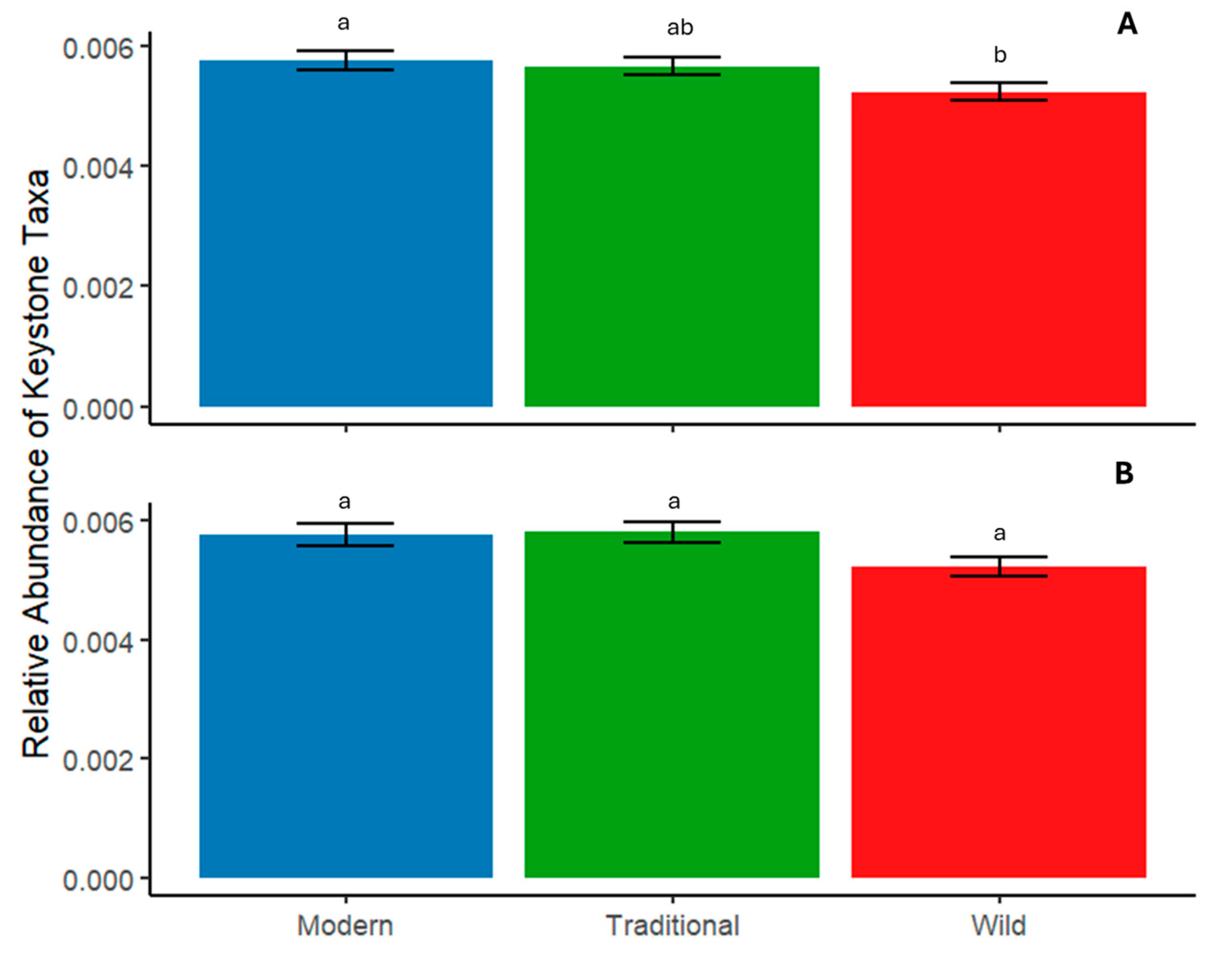
3.2. Tomato Domestication Influence on Soil Bacterial Interrelationships
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cordell, D.; Drangert, J.-O.; White, S. The story of phosphorus: Global food security and food for thought. Glob. Environ. Chang. 2009, 19, 292–305. [Google Scholar] [CrossRef]
- Ragot, S.A.; Kertesz, M.A.; Bünemann, E.K. phoD alkaline phosphatase gene diversity in soil. Appl. Environ. Microbiol. 2015, 81, 7281–7289. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Yuan, L.; Zhang, J.; Li, H.; Bai, Z.; Chen, X.; Zhang, W.; Zhang, F. Phosphorus dynamics: From soil to plant. Plant Physiol. 2011, 156, 997–1005. [Google Scholar] [CrossRef]
- Zhu, X.; Zhao, X.; Lin, Q.; Li, G. Distribution characteristics of phoD-harbouring bacterial community structure and its roles in phosphorus transformation in steppe soils in Northern China. J. Soil Sci. Plant Nutr. 2021, 21, 1531–1541. [Google Scholar] [CrossRef]
- Meyer, J.B.; Frapolli, M.; Keel, C.; Maurhofer, M. Pyrroloquinoline quinone biosynthesis gene pqqC, a novel molecular marker for studying the phylogeny and diversity of phosphate-solubilizing pseudomonads. Appl. Environ. Microbiol. 2011, 77, 7345–7354. [Google Scholar] [CrossRef]
- Magnusson, O.T.; Toyama, H.; Saeki, M.; Rojas, A.; Reed, J.C.; Liddington, R.C.; Klinman, J.P.; Schwarzenbacher, R. Quinone biogenesis: Structure and mechanism of PqqC, the final catalyst in the production of pyrroloquinoline quinone. Proc. Natl. Acad. Sci. USA 2004, 101, 7913–7918. [Google Scholar] [CrossRef] [PubMed]
- Yahya, M.; Islam, E.u.; Rasul, M.; Farooq, I.; Mahreen, N.; Tawab, A.; Irfan, M.; Rajput, L.; Amin, I.; Yasmin, S. Differential root exudation and architecture for improved growth of wheat mediated by phosphate solubilizing bacteria. Front. Microbiol. 2021, 12, 744094. [Google Scholar] [CrossRef]
- Beauregard, P.B. Not just sweet talkers: How roots stimulate their colonization by beneficial bacteria. In Advances in botanical Research; Elsevier: Amsterdam, The Netherlands, 2015; Volume 75, pp. 1–20. [Google Scholar]
- Berendsen, R.L.; Pieterse, C.M.; Bakker, P.A. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Berg, G.; Smalla, K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol. Ecol. 2009, 68, 1–13. [Google Scholar] [CrossRef]
- Afkairin, A.; Dixon, M.M.; Buchanan, C.; Ippolito, J.A.; Manter, D.K.; Davis, J.G.; Vivanco, J.M. Harnessing Phosphorous (P) Fertilizer-Insensitive Bacteria to Enhance Rhizosphere P Bioavailability in Legumes. Microorganisms 2024, 12, 353. [Google Scholar] [CrossRef]
- Dixon, M.M.; Afkairin, A.; Davis, J.G.; Chitwood-Brown, J.; Buchanan, C.M.; Ippolito, J.A.; Manter, D.K.; Vivanco, J.M. Tomato domestication rather than subsequent breeding events reduces microbial associations related to phosphorus recovery. Sci. Rep. 2024, 14, 9934. [Google Scholar] [CrossRef] [PubMed]
- Prischl, M.; Hackl, E.; Pastar, M.; Pfeiffer, S.; Sessitsch, A. Genetically modified Bt maize lines containing cry3Bb1, cry1A105 or cry1Ab2 do not affect the structure and functioning of root-associated endophyte communities. Appl. Soil Ecol. 2012, 54, 39–48. [Google Scholar] [CrossRef]
- Qiao, Q.; Wang, F.; Zhang, J.; Chen, Y.; Zhang, C.; Liu, G.; Zhang, H.; Ma, C.; Zhang, J. The variation in the rhizosphere microbiome of cotton with soil type, genotype and developmental stage. Sci. Rep. 2017, 7, 3940. [Google Scholar] [CrossRef]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, A.K.; Yin, C.; Hulbert, S.H. Community structure, species variation, and potential functions of rhizosphere-associated bacteria of different winter wheat (Triticum aestivum) cultivars. Front. Plant Sci. 2017, 8, 241964. [Google Scholar] [CrossRef]
- Abdallah, R.A.B.; Chikh-Rouhou, H.; Jabnoun-Khiareddine, H.; Sta-Baba, R.; Daami-Remadi, M. Fungal and bacterial rhizosphere microbiome associated with selected melon and snake melon genotypes. J. Microbiol. Biotechnol. Food Sci. 2021, 11, e4004. [Google Scholar]
- Sousa, R.M.S.; Mendes, L.W.; Antunes, J.E.L.; de Souza Oliveira, L.M.; Sousa, A.M.d.C.B.; Gomes, R.L.F.; de Almeida Lopes, A.C.; Araujo, F.F.; Melo, V.M.M.; Araujo, A.S.F. Diversity and structure of bacterial community in rhizosphere of lima bean. Appl. Soil Ecol. 2020, 150, 103490. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; Mendes, R.; Raaijmakers, J.M. Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant Mol. Biol. 2016, 90, 635–644. [Google Scholar] [CrossRef]
- Kovacs, E.; Rusu, T.; Lech, W.; Kovacs, M.; Roman, C. Rhizosphere microbiota profile changes with different genetic types of tomato species. Agric. -Rev. Știință Pract. Agric. 2019, 28, 140–150. [Google Scholar]
- Cordovez, V.; Rotoni, C.; Dini-Andreote, F.; Oyserman, B.; Carrión, V.J.; Raaijmakers, J.M. Successive plant growth amplifies genotype-specific assembly of the tomato rhizosphere microbiome. Sci. Total Environ. 2021, 772, 144825. [Google Scholar] [CrossRef]
- Demirer, G.S.; Gibson, D.J.; Yue, X.; Pan, K.; Elishav, E.; Khandal, H.; Horev, G.; Tarkowská, D.; Cantó-Pastor, A.; Kong, S. Phosphate deprivation-induced changes in tomato are mediated by an interaction between brassinosteroid signaling and zinc. New Phytol. 2023, 239, 1368–1383. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, Y.-F.; Li, Y.-S.; Jian, J.; Lian, T.-X. The rhizospheric microbiome becomes more diverse with maize domestication and genetic improvement. J. Integr. Agric. 2022, 21, 1188–1202. [Google Scholar] [CrossRef]
- Jaiswal, A.K.; Mengiste, T.D.; Myers, J.R.; Egel, D.S.; Hoagland, L.A. Tomato domestication attenuated responsiveness to a beneficial soil microbe for plant growth promotion and induction of systemic resistance to foliar pathogens. Front. Microbiol. 2020, 11, 604566. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, J.; Ingwell, L.L.; Li, X.; Kaplan, I. Domesticated tomatoes are more vulnerable to negative plant–soil feedbacks than their wild relatives. J. Ecol. 2019, 107, 1753–1766. [Google Scholar] [CrossRef]
- Yu, J.; Wang, L.; Jia, X.; Wang, Z.; Yu, X.; Ren, S.; Yang, Y.; Ye, X.; Wu, X.; Yi, K. Different microbial assembly between cultivated and wild tomatoes under P stress. Soil Sci. Environ. 2023, 2, 10. [Google Scholar] [CrossRef]
- Pantigoso, H.A.; Manter, D.K.; Vivanco, J.M. Phosphorus addition shifts the microbial community in the rhizosphere of blueberry (Vaccinium corymbosum L.). Rhizosphere 2018, 7, 1–7. [Google Scholar] [CrossRef]
- Cheng, H.; Yuan, M.; Duan, Q.; Sun, R.; Shen, Y.; Yu, Q.; Li, S. Influence of phosphorus fertilization patterns on the bacterial community in upland farmland. Ind. Crops Prod. 2020, 155, 112761. [Google Scholar] [CrossRef]
- Eltlbany, N.; Baklawa, M.; Ding, G.-C.; Nassal, D.; Weber, N.; Kandeler, E.; Neumann, G.; Ludewig, U.; Van Overbeek, L.; Smalla, K. Enhanced tomato plant growth in soil under reduced P supply through microbial inoculants and microbiome shifts. FEMS Microbiol. Ecol. 2019, 95, fiz124. [Google Scholar] [CrossRef]
- Yang, J.; Kloepper, J.W.; Ryu, C.-M. Rhizosphere bacteria help plants tolerate abiotic stress. Trends Plant Sci. 2009, 14, 1–4. [Google Scholar] [CrossRef]
- Tchakounté, G.V.T.; Berger, B.; Patz, S.; Becker, M.; Fankem, H.; Taffouo, V.D.; Ruppel, S. Selected rhizosphere bacteria help tomato plants cope with combined phosphorus and salt stresses. Microorganisms 2020, 8, 1844. [Google Scholar] [CrossRef]
- Gao, C.; Xu, L.; Montoya, L.; Madera, M.; Hollingsworth, J.; Chen, L.; Purdom, E.; Singan, V.; Vogel, J.; Hutmacher, R.B. Co-occurrence networks reveal more complexity than community composition in resistance and resilience of microbial communities. Nat. Commun. 2022, 13, 3867. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.R. Estimation of Available Phosphorus in Soils by Extraction with Sodium Bicarbonate; US Department of Agriculture: Washington, DC, USA, 1954. [Google Scholar]
- Seed Germination Recommendations. 2023. Available online: https://tgrc.ucdavis.edu/seedgermination (accessed on 1 August 2023).
- Urbaniak, G.C.; Plous, S. Research Randomizer (Version 4.0). Available online: https://www.randomizer.org/ (accessed on 1 September 2023).
- Pantigoso, H.A.; Manter, D.K.; Vivanco, J.M. Differential effects of phosphorus fertilization on plant uptake and rhizosphere microbiome of cultivated and non-cultivated potatoes. Microb. Ecol. 2020, 80, 169–180. [Google Scholar] [CrossRef]
- Curry, K.D.; Wang, Q.; Nute, M.G.; Tyshaieva, A.; Reeves, E.; Soriano, S.; Wu, Q.; Graeber, E.; Finzer, P.; Mendling, W. Emu: Species-level microbial community profiling of full-length 16S rRNA Oxford Nanopore sequencing data. Nat. Methods 2022, 19, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef] [PubMed]
- Clauset, A.; Newman, M.E.; Moore, C. Finding community structure in very large networks. Phys. Rev. E 2004, 70, 066111. [Google Scholar] [CrossRef]
- Yon, G.G.V. Building, Importing, and Exporting GEXF Graph Files with rgexf. J. Open Source Softw. 2021, 6, 3456. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the International AAAI Conference on Web and Social Media, San Jose, CA, USA, 17–20 May 2009; pp. 361–362. [Google Scholar]
- Fruchterman, T.M.; Reingold, E.M. Graph drawing by force-directed placement. Softw. Pract. Exp. 1991, 21, 1129–1164. [Google Scholar] [CrossRef]
- Gan, Z.; Li, N.; Ma, Y.; Lu, H. Trust network visualization based on force-directed layout. In Proceedings of the 2013 10th Web Information System and Application Conference, Washington, DC, USA, 10–15 November 2013; pp. 199–204. [Google Scholar]
- Qiu, L.; Zhang, Q.; Zhu, H.; Reich, P.B.; Banerjee, S.; van der Heijden, M.G.; Sadowsky, M.J.; Ishii, S.; Jia, X.; Shao, M. Erosion reduces soil microbial diversity, network complexity and multifunctionality. ISME J. 2021, 15, 2474–2489. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Chang, J.; Sun, Y.; Tian, L.; Ji, L.; Luo, S.; Nasir, F.; Kuramae, E.E.; Tian, C. The structure of rhizosphere fungal communities of wild and domesticated rice: Changes in diversity and co-occurrence patterns. Front. Microbiol. 2021, 12, 610823. [Google Scholar] [CrossRef]
- Sun, Y.; Tian, L.; Chang, J.; Shi, S.; Zhang, J.; Xie, H.; Cai, Y.; Chen, D.; Kuramae, E.E.; van Veen, J.A. Rice domestication influences the composition and function of the rhizosphere bacterial chemotaxis systems. Plant Soil 2021, 466, 81–99. [Google Scholar] [CrossRef]
- Feng, H.; Fu, R.; Hou, X.; Lv, Y.; Zhang, N.; Liu, Y.; Xu, Z.; Miao, Y.; Krell, T.; Shen, Q. Chemotaxis of beneficial rhizobacteria to root exudates: The first step towards root–microbe rhizosphere interactions. Int. J. Mol. Sci. 2021, 22, 6655. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liao, L.; Ye, Z.; Liu, H.; Zhang, C.; Zhang, L.; Liu, G.; Wang, G. Different bacterial co-occurrence patterns and community assembly between rhizosphere and bulk soils under N addition in the plant–soil system. Plant Soil 2022, 471, 697–713. [Google Scholar] [CrossRef]
- Siles, J.A.; García-Sánchez, M.; Gómez-Brandón, M. Studying microbial communities through co-occurrence network analyses during processes of waste treatment and in organically amended soils: A Review. Microorganisms 2021, 9, 1165. [Google Scholar] [CrossRef]
- Schouten, H.J.; Tikunov, Y.; Verkerke, W.; Finkers, R.; Bovy, A.; Bai, Y.; Visser, R.G. Breeding has increased the diversity of cultivated tomato in the Netherlands. Front. Plant Sci. 2019, 10, 500236. [Google Scholar] [CrossRef] [PubMed]
- Zsögön, A.; Čermák, T.; Naves, E.R.; Notini, M.M.; Edel, K.H.; Weinl, S.; Freschi, L.; Voytas, D.F.; Kudla, J.; Peres, L.E.P. De novo domestication of wild tomato using genome editing. Nat. Biotechnol. 2018, 36, 1211–1216. [Google Scholar] [CrossRef]
- Corrado, G.; Piffanelli, P.; Caramante, M.; Coppola, M.; Rao, R. SNP genotyping reveals genetic diversity between cultivated landraces and contemporary varieties of tomato. Bmc Genom. 2013, 14, 1–14. [Google Scholar] [CrossRef]
- Blanca, J.; Montero-Pau, J.; Sauvage, C.; Bauchet, G.; Illa, E.; Díez, M.J.; Francis, D.; Causse, M.; Van der Knaap, E.; Cañizares, J. Genomic variation in tomato, from wild ancestors to contemporary breeding accessions. BMC Genom. 2015, 16, 257. [Google Scholar] [CrossRef]
- Lee, S.A.; Kim, Y.; Kim, J.M.; Chu, B.; Joa, J.-H.; Sang, M.K.; Song, J.; Weon, H.-Y. A preliminary examination of bacterial, archaeal, and fungal communities inhabiting different rhizocompartments of tomato plants under real-world environments. Sci. Rep. 2019, 9, 9300. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; Carrión, V.J.; de Hollander, M.; Raaijmakers, J.M. The wild side of plant microbiomes. Microbiome 2018, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Jaramillo, J.E.; Carrión, V.J.; Bosse, M.; Ferrão, L.F.; De Hollander, M.; Garcia, A.A.; Ramírez, C.A.; Mendes, R.; Raaijmakers, J.M. Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits. ISME J. 2017, 11, 2244–2257. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Garrido-Oter, R.; Münch, P.C.; Weiman, A.; Dröge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef]
- Smulders, L.; Benítez, E.; Moreno, B.; López-García, Á.; Pozo, M.J.; Ferrero, V.; de la Peña, E.; Alcalá Herrera, R. Tomato domestication affects potential functional molecular pathways of root-associated soil bacteria. Plants 2021, 10, 1942. [Google Scholar] [CrossRef]
- Yue, H.; Yue, W.; Jiao, S.; Kim, H.; Lee, Y.-H.; Wei, G.; Song, W.; Shu, D. Plant domestication shapes rhizosphere microbiome assembly and metabolic functions. Microbiome 2023, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.; Rohrbaugh, C.; Afkairin, A.; Vivanco, J. Impacts of the Green Revolution on Rhizosphere Microbiology Related to Nutrient Acquisition. Appl. Microbiol. 2022, 2, 992–1003. [Google Scholar] [CrossRef]
- Lucero, M.; Debolt, S.; Unc, A.; Ruiz-Font, A.; Reyes, L.; McCulley, R.L.; Alderman, S.; Dinkins, R.; Barrow, J.; Samac, D. Using microbial community interactions within plant microbiomes to advance an evergreen agricultural revolution. In Sustainable Agroecosystems in Climate Change Mitigation; Wageningen Academic: Wageningen, The Netherlands, 2014; pp. 183–202. [Google Scholar]
- Schmidt, J.E.; Kent, A.D.; Brisson, V.L.; Gaudin, A.C. Agricultural management and plant selection interactively affect rhizosphere microbial community structure and nitrogen cycling. Microbiome 2019, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Sardans, J.; Sun, D.; Yan, R.; Wu, H.; Ni, R.; Peñuelas, J. Microbial diversity and keystone species drive soil nutrient cycling and multifunctionality following mangrove restoration. Environ. Res. 2024, 251, 118715. [Google Scholar] [CrossRef]
- Bruto, M.; Prigent-Combaret, C.; Muller, D.; Moënne-Loccoz, Y. Analysis of genes contributing to plant-beneficial functions in plant growth-promoting rhizobacteria and related Proteobacteria. Sci. Rep. 2014, 4, 6261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ullah, F.; Ahmad, R.; Shah, S.U.A.; Khan, A.; Adnan, M. Response of soil proteobacteria to biochar amendment in sustainable agriculture-a mini review. J. Soil Plant Environ. 2022, 1, 16–30. [Google Scholar] [CrossRef]
- Gebauer, L.; Breitkreuz, C.; Heintz-Buschart, A.; Reitz, T.; Buscot, F.; Tarkka, M.; Bouffaud, M.-L. Water deficit history selects plant beneficial soil bacteria differently under conventional and organic farming. Front. Microbiol. 2022, 13, 824437. [Google Scholar] [CrossRef]
- Shi, S.; Nuccio, E.E.; Shi, Z.J.; He, Z.; Zhou, J.; Firestone, M.K. The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol. Lett. 2016, 19, 926–936. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dixon, M.M.; Afkairin, A.; Manter, D.K.; Vivanco, J. Rhizosphere Microbiome Co-Occurrence Network Analysis across a Tomato Domestication Gradient. Microorganisms 2024, 12, 1756. https://doi.org/10.3390/microorganisms12091756
Dixon MM, Afkairin A, Manter DK, Vivanco J. Rhizosphere Microbiome Co-Occurrence Network Analysis across a Tomato Domestication Gradient. Microorganisms. 2024; 12(9):1756. https://doi.org/10.3390/microorganisms12091756
Chicago/Turabian StyleDixon, Mary M., Antisar Afkairin, Daniel K. Manter, and Jorge Vivanco. 2024. "Rhizosphere Microbiome Co-Occurrence Network Analysis across a Tomato Domestication Gradient" Microorganisms 12, no. 9: 1756. https://doi.org/10.3390/microorganisms12091756
APA StyleDixon, M. M., Afkairin, A., Manter, D. K., & Vivanco, J. (2024). Rhizosphere Microbiome Co-Occurrence Network Analysis across a Tomato Domestication Gradient. Microorganisms, 12(9), 1756. https://doi.org/10.3390/microorganisms12091756