
Chikungunya Virus RNA Secondary Structures Impact Defective Viral Genome Production

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Cloning Selected Disrupted Secondary Structure (D2S) Mutant
2.3. Plaque Assay
2.4. Viral Passages
2.5. Growth Curves
2.6. Deep-Sequencing
2.7. SHAPE-MaP of WT and D2S CHIKV RNA
2.8. Data Analysis
2.8.1. Alignment and Identification of DVGs
2.8.2. Correlation between DVG Nucleotide Allele Frequency and SHAPE Reactivity
2.8.3. Comparison of the SHAPE Reactivity of Nucleotides Used or Not Used as DVG Breakpoints
2.8.4. Comparison of DGVs Generated in WT and D2S Mutant
2.8.5. DVG Entropy
2.8.6. Statistical Analysis
3. Results
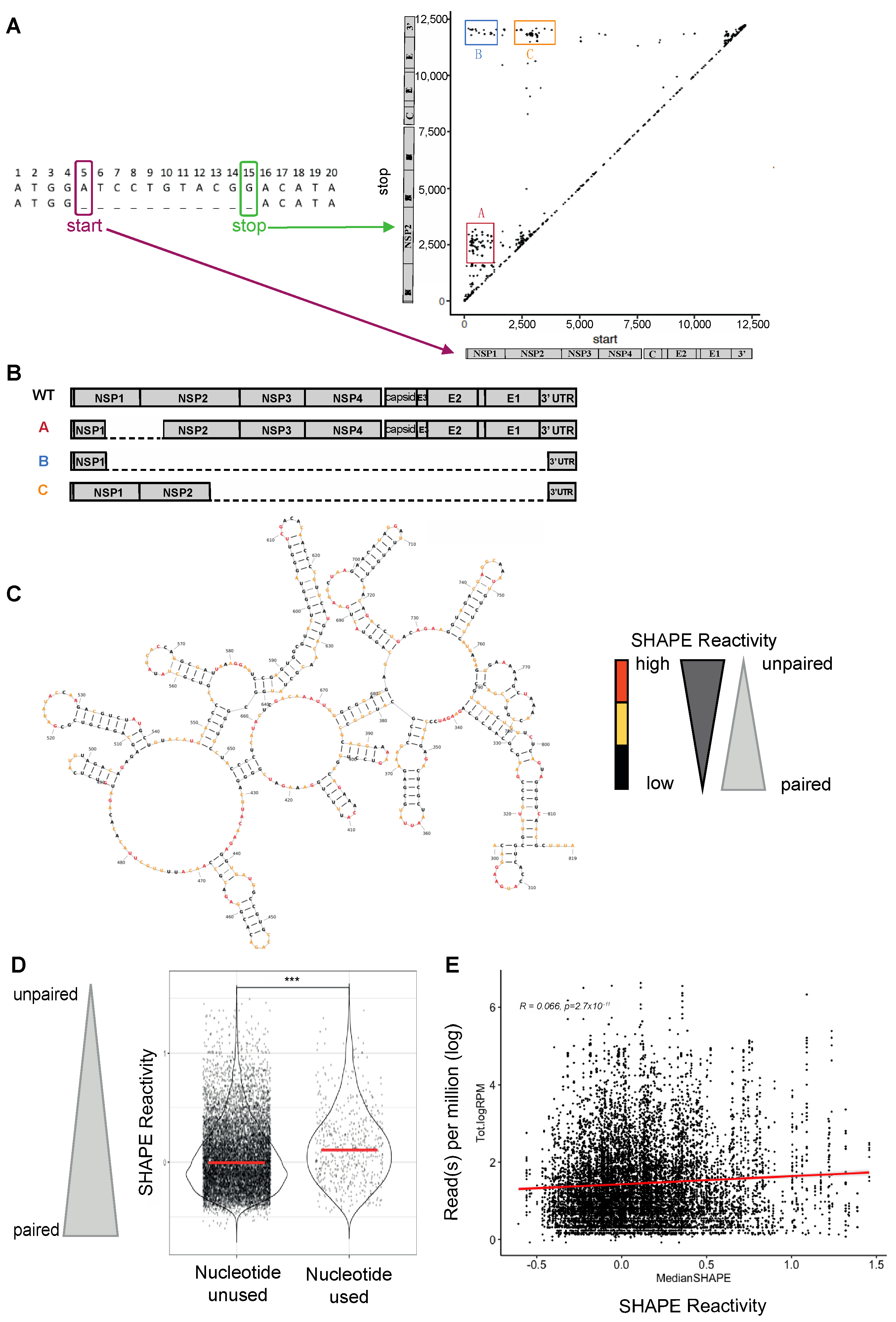
3.1. Preferential Generation of DVG Breakpoints at Unpaired Nucleotides
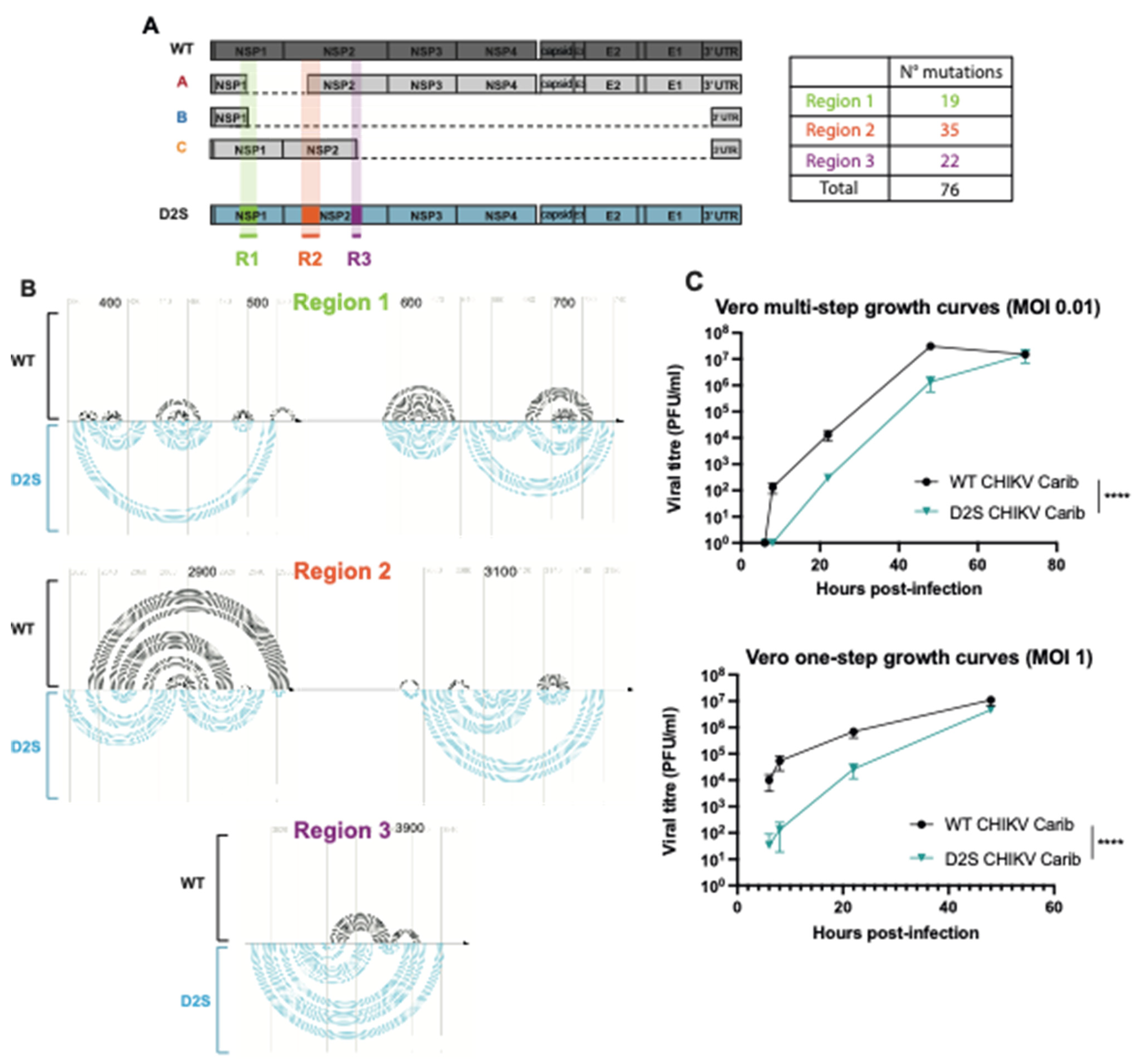
3.2. CHIKV-D2S Mutant with Disrupted Secondary Structure Is Viable in Cell Culture
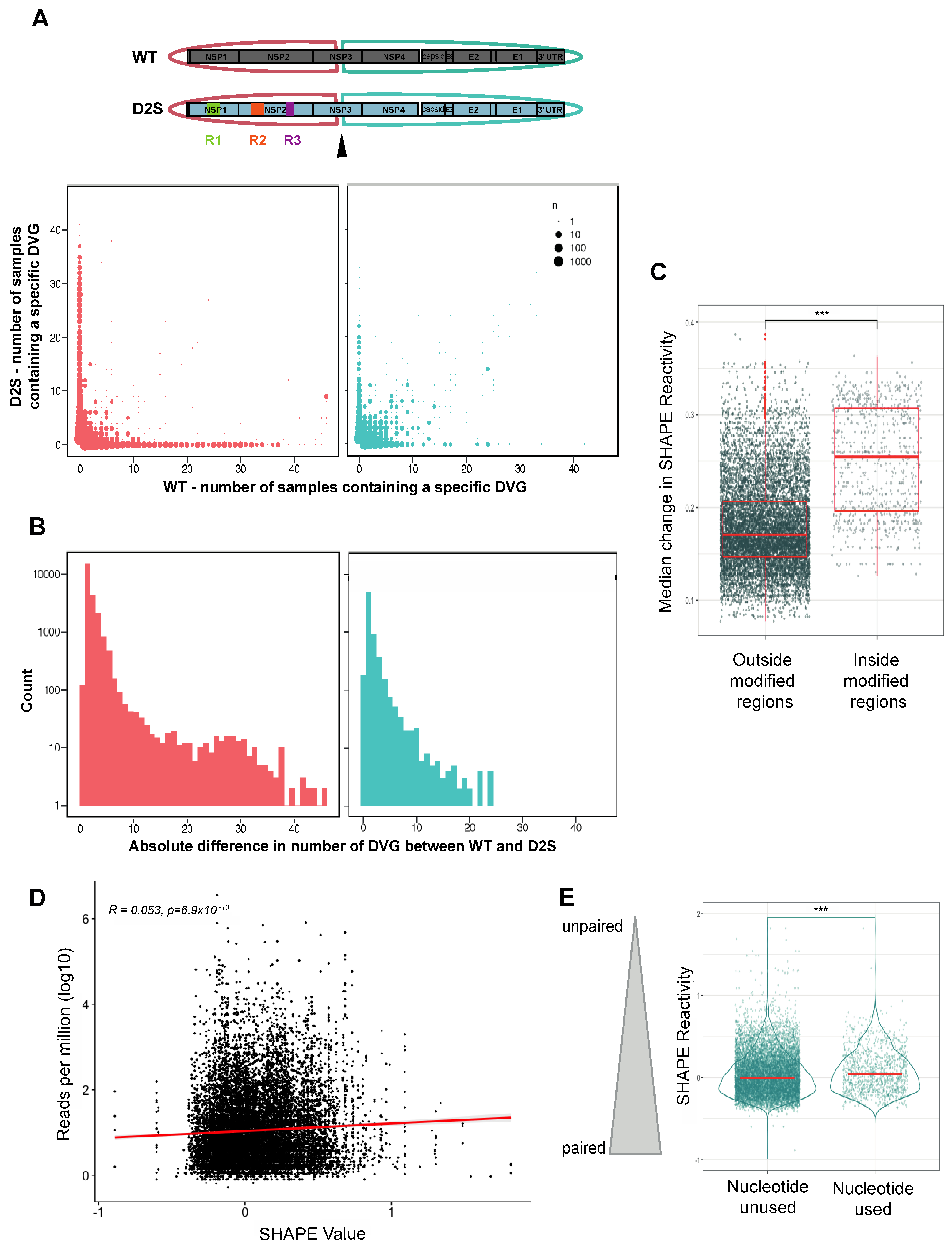
3.3. Altered DVG Generation in CHIKV D2S, Compared to WT Virus
3.4. DVG Generation Is Governed by RNA Secondary Structures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Borgherini, G.; Poubeau, P.; Staikowsky, F.; Lory, M.; Moullec, N.L.; Becquart, J.P.; Wengling, C.; Michault, A.; Paganin, F. Outbreak of Chikungunya on Reunion Island: Early Clinical and Laboratory Features in 157 Adult Patients. Clin. Infect. Dis. 2007, 44, 1401–1407. [Google Scholar] [CrossRef]
- Suhrbier, A.; Jaffar-Bandjee, M.-C.; Gasque, P. Arthritogenic Alphaviruses—An Overview. Nature Reviews. Rheumatology 2012, 8, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Bouquillard, E.; Fianu, A.; Bangil, M.; Charlette, N.; Ribéra, A.; Michault, A.; Favier, F.; Simon, F.; Flipo, R.-M. Rheumatic Manifestations Associated with Chikungunya Virus Infection: A Study of 307 Patients with 32-Month Follow-up (RHUMATOCHIK Study). Jt. Bone Spine Rev. Rhum. 2018, 85, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Levi, L.I.; Vignuzzi, M. Arthritogenic Alphaviruses: A Worldwide Emerging Threat? Microorganisms 2019, 7, 133. [Google Scholar] [CrossRef] [PubMed]
- Couturier, E.; Guillemin, F.; Mura, M.; Léon, L.; Virion, J.-M.; Letort, M.-J.; De Valk, H.; Simon, F.; Vaillant, V. Impaired Quality of Life after Chikungunya Virus Infection: A 2-Year Follow-up Study. Rheumatology 2012, 51, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Borgherini, G.; Poubeau, P.; Jossaume, A.; Gouix, A.; Cotte, L.; Michault, A.; Arvin-Berod, C.; Paganin, F. Persistent Arthralgia Associated with Chikungunya Virus: A Study of 88 Adult Patients on Reunion Island. Clin. Infect. Dis. 2008, 47, 469–475. [Google Scholar] [CrossRef]
- Chretien, J.-P.; Anyamba, A.; Bedno, S.A.; Breiman, R.F.; Sang, R.; Sergon, K.; Powers, A.M.; Onyango, C.O.; Small, J.; Tucker, C.J.; et al. Drought-Associated Chikungunya Emergence Along Coastal East Africa. Am. J. Trop. Med. Hyg. 2007, 76, 405–407. [Google Scholar] [CrossRef]
- Vignuzzi, M.; Higgs, S. The Bridges and Blockades to Evolutionary Convergence on the Road to Predicting Chikungunya Virus Evolution. Annu. Rev. Virol. 2017, 4, 181–200. [Google Scholar] [CrossRef]
- Poirier, E.Z.; Goic, B.; Tomé-Poderti, L.; Frangeul, L.; Boussier, J.; Gausson, V.; Blanc, H.; Vallet, T.; Loyd, H.; Levi, L.I.; et al. Dicer-2-Dependent Generation of Viral DNA from Defective Genomes of RNA Viruses Modulates Antiviral Immunity in Insects. Cell Host Microbe 2018, 23, 353–365.e8. [Google Scholar] [CrossRef]
- Levi, L.I.; Rezelj, V.V.; Henrion-Lacritick, A.; Erazo, D.; Boussier, J.; Vallet, T.; Bernhauerová, V.; Suzuki, Y.; Carrau, L.; Weger-Lucarelli, J.; et al. Defective Viral Genomes from Chikungunya Virus Are Broad-Spectrum Antivirals and Prevent Virus Dissemination in Mosquitoes. PLoS Pathog. 2021, 17, e1009110. [Google Scholar] [CrossRef]
- Vignuzzi, M.; López, C.B. Defective Viral Genomes Are Key Drivers of the Virus–Host Interaction. Nat. Microbiol. 2019, 4, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Rezelj, V.V.; Levi, L.I.; Vignuzzi, M. The Defective Component of Viral Populations. Curr. Opin. Virol. 2018, 33, 74–80. [Google Scholar] [CrossRef]
- Monroe, S.S.; Schlesinger, S. Common and Distinct Regions of Defective-Interfering RNAs of Sindbis Virus. J. Virol. 1984, 49, 865–872. [Google Scholar] [CrossRef]
- Marcus, P.I.; Sekellick, M.J. Defective Interfering Particles with Covalently Linked [+/-]RNA Induce Interferon. Nature 1977, 266, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jain, D.; Koziol-White, C.J.; Genoyer, E.; Gilbert, M.; Tapia, K.; Panettieri, R.A.; Hodinka, R.L.; López, C.B. Immunostimulatory Defective Viral Genomes from Respiratory Syncytial Virus Promote a Strong Innate Antiviral Response during Infection in Mice and Humans. PLoS Pathog. 2015, 11, e1005122. [Google Scholar] [CrossRef] [PubMed]
- Vasilijevic, J.; Zamarreño, N.; Oliveros, J.C.; Rodriguez-Frandsen, A.; Gómez, G.; Rodriguez, G.; Pérez-Ruiz, M.; Rey, S.; Barba, I.; Pozo, F.; et al. Reduced Accumulation of Defective Viral Genomes Contributes to Severe Outcome in Influenza Virus Infected Patients. PLoS Pathog. 2017, 13, e1006650. [Google Scholar] [CrossRef]
- Easton, A.J.; Scott, P.D.; Edworthy, N.L.; Meng, B.; Marriott, A.C.; Dimmock, N.J. A Novel Broad-Spectrum Treatment for Respiratory Virus Infections: Influenza-Based Defective Interfering Virus Provides Protection against Pneumovirus Infection In Vivo. Vaccine 2011, 29, 2777–2784. [Google Scholar] [CrossRef]
- Rezelj, V.V.; Carrau, L.; Merwaiss, F.; Levi, L.I.; Erazo, D.; Tran, Q.D.; Henrion-Lacritick, A.; Gausson, V.; Suzuki, Y.; Shengjuler, D.; et al. Defective Viral Genomes as Therapeutic Interfering Particles against Flavivirus Infection in Mammalian and Mosquito Hosts. Nat. Commun. 2021, 12, 2290. [Google Scholar] [CrossRef]
- Wang, H.; Cui, X.; Cai, X.; An, T. Recombination in Positive-Strand RNA Viruses. Front. Microbiol. 2022, 13, 870759. [Google Scholar] [CrossRef]
- Nicholson, B.L.; White, K.A. Exploring the Architecture of Viral RNA Genomes. Curr. Opin. Virol. 2015, 12, 66–74. [Google Scholar] [CrossRef]
- Rausch, J.W.; Sztuba-Solinska, J.; Le Grice, S.F.J. Probing the Structures of Viral RNA Regulatory Elements with SHAPE and Related Methodologies. Front. Microbiol. 2017, 8, 2634. [Google Scholar] [CrossRef]
- Nicholson, B.L.; White, K.A. Functional Long-Range RNA-RNA Interactions in Positive-Strand RNA Viruses. Nat. Rev. Microbiol. 2014, 12, 493–504. [Google Scholar] [CrossRef]
- Kutchko, K.M.; Madden, E.A.; Morrison, C.; Plante, K.S.; Sanders, W.; Vincent, H.A.; Cisneros, M.C.C.; Long, K.M.; Moorman, N.J.; Heise, M.T.; et al. Structural divergence creates new functional features in alphavirus genomes. Nucleic Acids Res. 2018, 46, 3657–3670. [Google Scholar] [CrossRef] [PubMed]
- Galetto, R.; Moumen, A.; Giacomoni, V.; Véron, M.; Charneau, P.; Negroni, M. The Structure of HIV-1 Genomic RNA in the Gp120 Gene Determines a Recombination Hot Spot In Vivo. J. Biol. Chem. 2004, 279, 36625–36632. [Google Scholar] [CrossRef] [PubMed]
- Moumen, A.; Polomack, L.; Unge, T.; Véron, M.; Buc, H.; Negroni, M. Evidence for a Mechanism of Recombination during Reverse Transcription Dependent on the Structure of the Acceptor RNA. J. Biol. Chem. 2003, 278, 15973–15982. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Martin, D.P.; Weeks, K.M.; Negroni, M. RNA Structures Facilitate Recombination-Mediated Gene Swapping in HIV-1. J. Virol. 2010, 84, 12675–12682. [Google Scholar] [CrossRef] [PubMed]
- Figlerowicz, M. Role of RNA structure in non-homologous recombination between genomic molecules of brome mosaic virus. Nucleic Acids Res. 2000, 28, 1714–1723. [Google Scholar] [CrossRef]
- Dedepsidis, E.; Kyriakopoulou, Z.; Pliaka, V.; Markoulatos, P. Correlation between Recombination Junctions and RNA Secondary Structure Elements in Poliovirus Sabin Strains. Virus Genes 2010, 41, 181–191. [Google Scholar] [CrossRef]
- Chao, M.; Wang, T.-C.; Lin, C.-C.; Wang, R.Y.-L.; Lin, W.-B.; Lee, S.-E.; Cheng, Y.-Y.; Yeh, C.-T.; Iang, S.-B. Analyses of a Whole-Genome Inter-Clade Recombination Map of Hepatitis Delta Virus Suggest a Host Polymerase-Driven and Viral RNA Structure-Promoted Template-Switching Mechanism for Viral RNA Recombination. Oncotarget 2017, 8, 60841–60859. [Google Scholar] [CrossRef]
- Havelda, Z.; Burgyán, J.; Dalmay, T. Secondary Structure-Dependent Evolution of Cymbidium Ringspot Virus Defective Interfering RNA. J. Gen. Virol. 1997, 78, 1227–1234. [Google Scholar] [CrossRef]
- Merino, E.J.; Wilkinson, K.A.; Coughlan, J.L.; Weeks, K.M. RNA Structure Analysis at Single Nucleotide Resolution by Selective 2′-Hydroxyl Acylation and Primer Extension (SHAPE). J. Am. Chem. Soc. 2005, 127, 4223–4231. [Google Scholar] [CrossRef]
- Low, J.T.; Weeks, K.M. SHAPE-Directed RNA Secondary Structure Prediction. Methods 2010, 52, 150–158. [Google Scholar] [CrossRef]
- Wilkinson, K.A.; Merino, E.J.; Weeks, K.M. Selective 2′-Hydroxyl Acylation Analyzed by Primer Extension (SHAPE): Quantitative RNA Structure Analysis at Single Nucleotide Resolution. Nat. Protoc. 2006, 1, 1610–1616. [Google Scholar] [CrossRef]
- Siegfried, N.A.; Busan, S.; Rice, G.M.; Nelson, J.A.; Weeks, K.M. RNA Motif Discovery by SHAPE and Mutational Profiling (SHAPE-MaP). Nat. Methods 2014, 11, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Madden, E.A.; Plante, K.S.; Morrison, C.R.; Kutchko, K.M.; Sanders, W.; Long, K.M.; Taft-Benz, S.; Cisneros, M.C.C.; White, A.M.; Sarkar, S.; et al. Using SHAPE-MaP to Model RNA Secondary Structure and Identify 3′UTR Variation in Chikungunya Virus. Édité par Rebecca Ellis Dutch. J. Virol. 2020, 94, e00701–e00720. [Google Scholar] [CrossRef]
- Stapleford, K.A.; Moratorio, G.; Henningsson, R.; Chen, R.; Matheus, S.; Enfissi, A.; Weissglas-Volkov, D.; Isakov, O.; Blanc, H.; Mounce, B.C.; et al. Whole-Genome Sequencing Analysis from the Chikungunya Virus Caribbean Outbreak Reveals Novel Evolutionary Genomic Elements. PLoS Negl. Trop. Dis. 2016, 10, e0004402. [Google Scholar] [CrossRef]
- Jorge, D.M.d.M.; Mills, R.E.; Lauring, A.S. CodonShuffle: A Tool for Generating and Analyzing Synonymously Mutated Sequences. Virus Evol. 2015, 1, vev012. [Google Scholar] [CrossRef] [PubMed]
- Marinus, T.; Fessler, A.B.; Ogle, C.A.; Incarnato, D. A Novel SHAPE Reagent Enables the Analysis of RNA Structure in Living Cells with Unprecedented Accuracy. Nucleic Acids Res. 2021, 49, e34. [Google Scholar] [CrossRef] [PubMed]
- von Magnus, P. Incomplete Forms of Influenza Virus. Adv. Virus Res. 1954, 2, 59–79. [Google Scholar]
- von Magnus, P. Studies on Interference in Experimental Influenza. Arkiv för Kemi, Mineralogi och Geologi 24 (1). Available online: https://books.google.fr/books/about/Studies_on_interference_in_experimental.html?id=f-IEHQAACAAJ&redir_esc=y (accessed on 1 August 2024).
- Frolov, I.; Hardy, R.; Rice, C.M. Cis-Acting RNA Elements at the 5′ End of Sindbis Virus Genome RNA Regulate Minus- and plus-Strand RNA Synthesis. RNA 2001, 7, 1638–1651. [Google Scholar] [CrossRef]
- Niesters, H.G.; Strauss, J.H. Mutagenesis of the Conserved 51-Nucleotide Region of Sindbis Virus. J. Virol. 1990, 64, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.E.; Chung, B.Y.; Fleeton, M.N.; Atkins, J.F. Discovery of Frameshifting in Alphavirus 6K Resolves a 20-Year Enigma. Virol. J. 2008, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Tomezsko, P.J.; Corbin, V.D.A.; Gupta, P.; Swaminathan, H.; Glasgow, M.; Persad, S.; Edwards, M.D.; Mcintosh, L.; Papenfuss, A.T.; Emery, A.; et al. Determination of RNA Structural Diversity and Its Role in HIV-1 RNA Splicing. Nature 2020, 582, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Baird, H.A.; Galetto, R.; Gao, Y.; Simon-Loriere, E.; Abreha, M.; Archer, J.; Fan, J.; Robertson, D.L.; Arts, E.J.; Negroni, M. Sequence Determinants of Breakpoint Location during HIV-1 Intersubtype Recombination. Nucleic Acids Res. 2006, 34, 5203–5216. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Holmes, E.C. Why Do RNA Viruses Recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef]
- Smola, M.J.; Rice, G.M.; Busan, S.; Siegfried, N.A.; Weeks, K.M. Selective 2′-Hydroxyl Acylation Analyzed by Primer Extension and Mutational Profiling (SHAPE-MaP) for Direct, Versatile and Accurate RNA Structure Analysis. Nat. Protoc. 2015, 10, 1643–1669. [Google Scholar] [CrossRef]
- Huber, R.G.; Ni Lim, X.; Ng, W.C.; Sim, A.Y.L.; Poh, H.X.; Shen, Y.; Lim, S.Y.; Sundstrom, K.B.; Sun, X.; Aw, J.G.; et al. Structure Mapping of Dengue and Zika Viruses Reveals Functional Long-Range Interactions. Nat. Commun. 2019, 10, 1408. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levi, L.I.; Madden, E.A.; Boussier, J.; Erazo, D.; Sanders, W.; Vallet, T.; Bernhauerova, V.; Moorman, N.J.; Heise, M.T.; Vignuzzi, M. Chikungunya Virus RNA Secondary Structures Impact Defective Viral Genome Production. Microorganisms 2024, 12, 1794. https://doi.org/10.3390/microorganisms12091794
Levi LI, Madden EA, Boussier J, Erazo D, Sanders W, Vallet T, Bernhauerova V, Moorman NJ, Heise MT, Vignuzzi M. Chikungunya Virus RNA Secondary Structures Impact Defective Viral Genome Production. Microorganisms. 2024; 12(9):1794. https://doi.org/10.3390/microorganisms12091794
Chicago/Turabian StyleLevi, Laura I., Emily A. Madden, Jeremy Boussier, Diana Erazo, Wes Sanders, Thomas Vallet, Veronika Bernhauerova, Nathaniel J. Moorman, Mark T. Heise, and Marco Vignuzzi. 2024. "Chikungunya Virus RNA Secondary Structures Impact Defective Viral Genome Production" Microorganisms 12, no. 9: 1794. https://doi.org/10.3390/microorganisms12091794
APA StyleLevi, L. I., Madden, E. A., Boussier, J., Erazo, D., Sanders, W., Vallet, T., Bernhauerova, V., Moorman, N. J., Heise, M. T., & Vignuzzi, M. (2024). Chikungunya Virus RNA Secondary Structures Impact Defective Viral Genome Production. Microorganisms, 12(9), 1794. https://doi.org/10.3390/microorganisms12091794