
Temporal Investigation of the Maternal Origins of Fetal Gut Microbiota

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participant Sampling
2.2. Microbiome Sequencing
2.3. Data and Bioinformatic Analysis
3. Results
3.1. Sequencing Depth and Quality Control
3.2. Operational Taxonomic Unit (OTU) Clustering and Taxonomic Assignment
3.3. Microbial Diversity and Composition
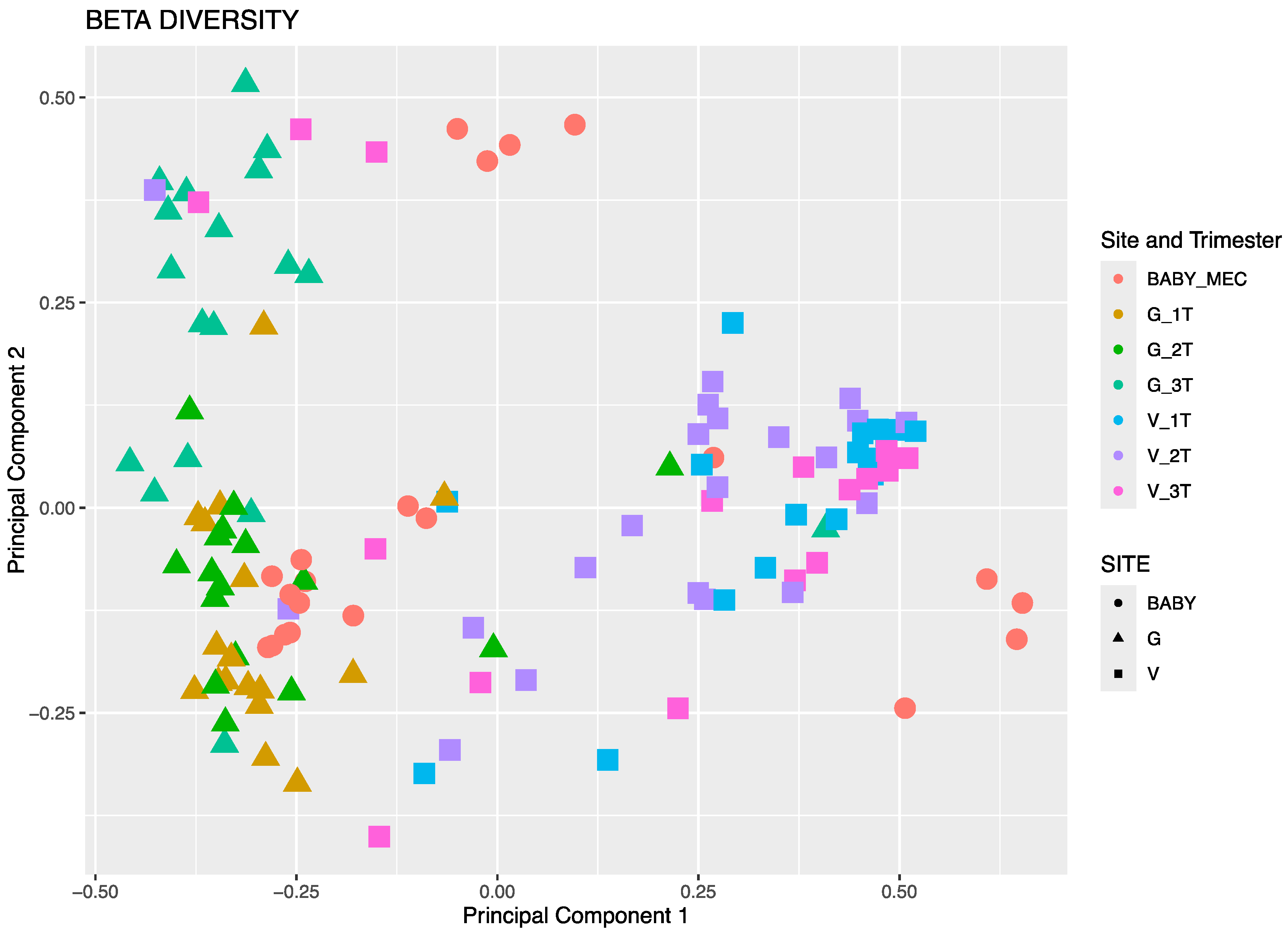
3.3.1. Beta Diversity by Principal Component Analysis
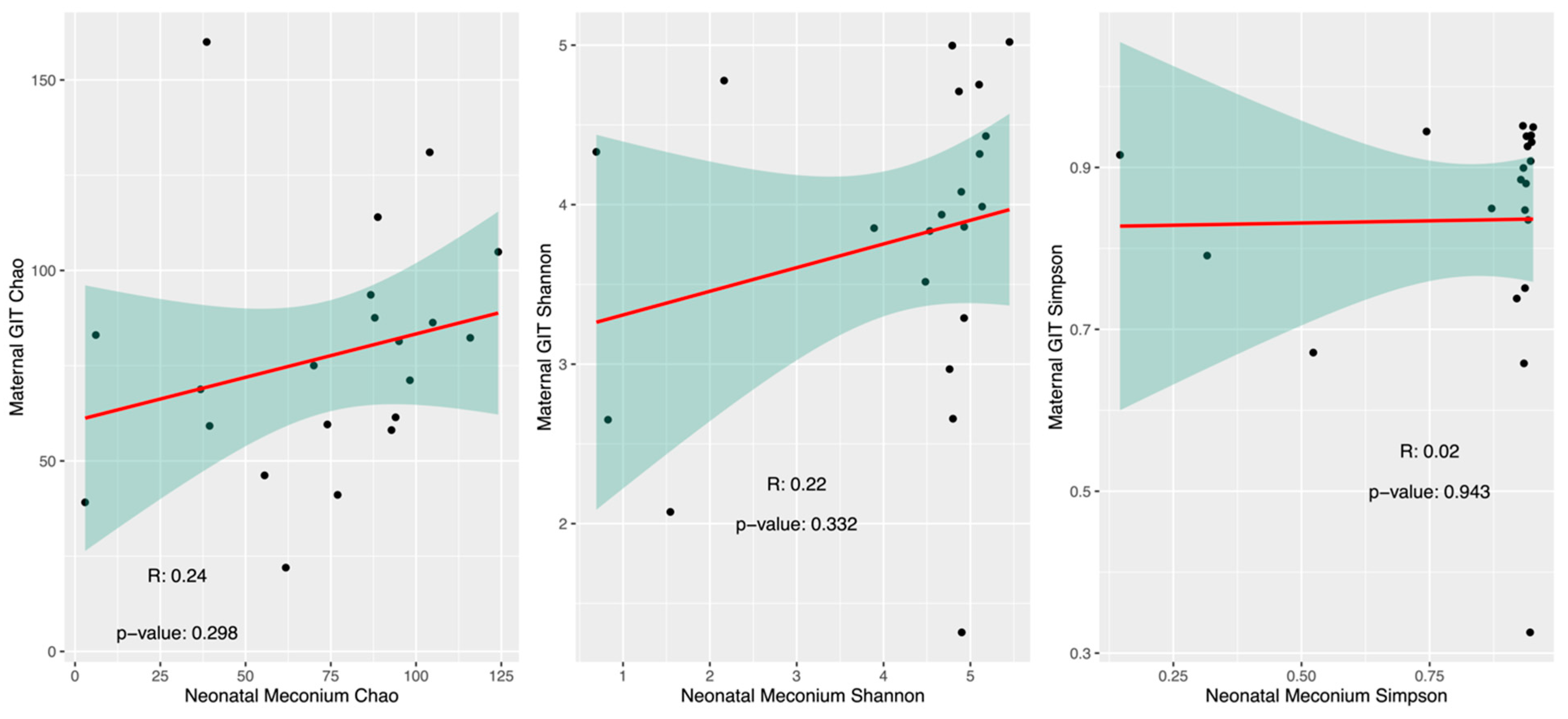
3.3.2. Alpha Diversity
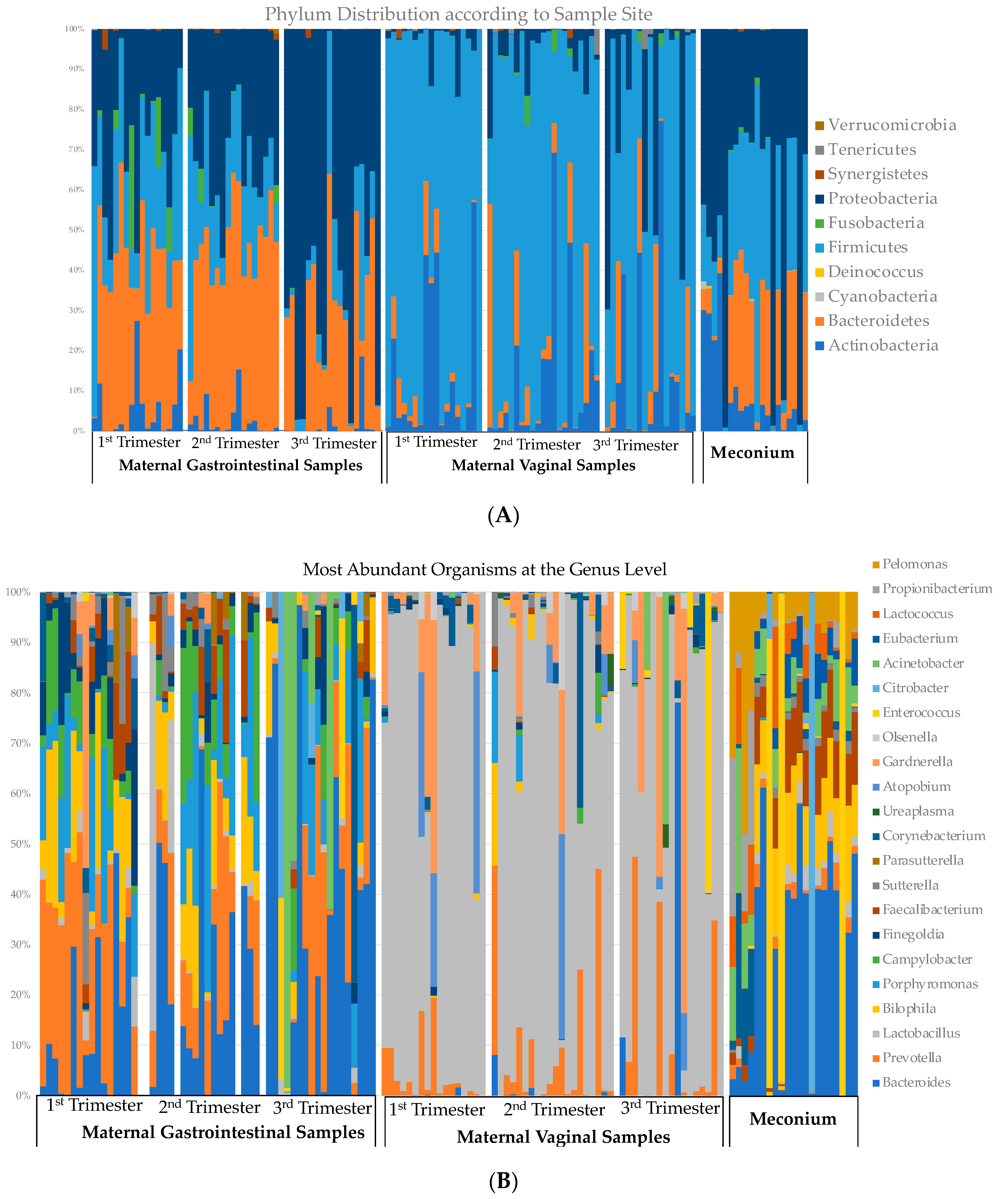
3.4. Relative Abundance across Gestation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jimenez, E.; Marin, M.L.; Martin, R.; Odriozola, J.M.; Olivares, M.; Xaus, J.; Fernandez, L.; Rodriguez, J.M. Is meconium from healthy newborns actually sterile? Res. Microbiol. 2008, 159, 187–193. [Google Scholar] [CrossRef]
- Funkhouser, L.J.; Bordenstein, S.R. Mom knows best: The universality of maternal microbial transmission. PLoS Biol. 2013, 11, e1001631. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.; Scott, K.P.; Khan, S.; Martin, J.C.; Berry, S.H.; Stevenson, M.; Okpapi, A.; Munro, M.J.; Hold, G.L. First-Pass Meconium Samples from Healthy Term Vaginally-Delivered Neonates: An Analysis of the Microbiota. PLoS ONE 2015, 10, e0133320. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.W.; Clemente, J.C.; Peter, I.; Loos, R.J.F. The prenatal gut microbiome: Are we colonized with bacteria in utero? Pediatr. Obes. 2017, 12 (Suppl. S1), 3–17. [Google Scholar] [CrossRef] [PubMed]
- Stinson, L.F.; Boyce, M.C.; Payne, M.S.; Keelan, J.A. The Not-so-Sterile Womb: Evidence That the Human Fetus Is Exposed to Bacteria Prior to Birth. Front. Microbiol. 2019, 10, 1124. [Google Scholar] [CrossRef]
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the human infant intestinal microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, e00036-17. [Google Scholar] [CrossRef]
- Ziętek, M.; Celewicz, Z.; Szczuko, M. Short-Chain Fatty Acids, Maternal Microbiota and Metabolism in Pregnancy. Nutrients 2021, 13, 1244. [Google Scholar] [CrossRef]
- Dong, T.; Chen, T.; White, R.A., 3rd; Wang, X.; Hu, W.; Liang, Y.; Zhang, Y.; Lu, C.; Chen, M.; Aase, H.; et al. Meconium microbiome associates with the development of neonatal jaundice. Clin. Transl. Gastroenterol. 2018, 9, 182. [Google Scholar] [CrossRef]
- Ding, J.; Ma, X.; Han, L.; Zhao, X.; Li, A.; Xin, Q.; Lian, W.; Li, Z.; Ren, H.; Ren, Z. Gut microbial alterations in neonatal jaundice pre- and post-treatment. Biosci. Rep. 2021, 41, BSR20210362. [Google Scholar] [CrossRef]
- Madan, J.C.; Salari, R.C.; Saxena, D.; Davidson, L.; O’Toole, G.A.; Moore, J.H.; Sogin, M.L.; Foster, J.A.; Edwards, W.H.; Palumbo, P.; et al. Gut microbial colonisation in premature neonates predicts neonatal sepsis. Arch. Dis. Child.-Fetal Neonatal Ed. 2012, 97, F456–F462. [Google Scholar] [CrossRef]
- Dornelles, L.V.; Procianoy, R.S.; Roesch, L.F.W.; Corso, A.L.; Dobbler, P.T.; Mai, V.; Silveira, R.C. Meconium microbiota predicts clinical early-onset neonatal sepsis in preterm neonates. J. Matern.-Fetal Neonatal Med. 2020, 35, 1935–1943. [Google Scholar] [CrossRef]
- Terrazzan Nutricionist, A.C.; Procianoy, R.S.; Roesch, L.F.W.; Corso, A.L.; Dobbler, P.T.; Silveira, R.C. Meconium microbiome and its relation to neonatal growth and head circumference catch-up in preterm infants. PLoS ONE 2020, 15, e0238632. [Google Scholar] [CrossRef] [PubMed]
- Ardissone, A.N.; de la Cruz, D.M.; Davis-Richardson, A.G.; Rechcigl, K.T.; Li, N.; Drew, J.C.; Murgas-Torrazza, R.; Sharma, R.; Hudak, M.L.; Triplett, E.W.; et al. Meconium microbiome analysis identifies bacteria correlated with premature birth. PLoS ONE 2014, 9, e90784. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.; Moles, L.; Espinosa-Martos, I.; Bustos, G.; de Vos, W.M.; Fernandez, L.; Rodriguez, J.M.; Fuentes, S.; Jimenez, E. Bacteriological and Immunological Profiling of Meconium and Fecal Samples from Preterm Infants: A Two-Year Follow-Up Study. Nutrients 2017, 9, 1293. [Google Scholar] [CrossRef]
- DiGiulio, D.B.; Romero, R.; Kusanovic, J.P.; Gómez, R.; Kim, C.J.; Seok, K.S.; Gotsch, F.; Mazaki-Tovi, S.; Vaisbuch, E.; Sanders, K.; et al. Prevalence and diversity of microbes in the amniotic fluid, the fetal inflammatory response, and pregnancy outcome in women with preterm pre-labor rupture of membranes. Am. J. Reprod. Immunol. 2010, 64, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Megli, C.J.; Coyne, C.B. Infections at the maternal-fetal interface: An overview of pathogenesis and defence. Nat. Rev. Microbiol. 2022, 20, 67–82. [Google Scholar] [CrossRef]
- Mishra, A.; Lai, G.C.; Yao, L.J.; Aung, T.T.; Shental, N.; Rotter-Maskowitz, A.; Shepherdson, E.; Singh, G.S.N.; Pai, R.; Shanti, A.; et al. Microbial exposure during early human development primes fetal immune cells. Cell 2021, 184, 3394–3409. [Google Scholar] [CrossRef]
- Daskalakis, G.; Psarris, A.; Koutras, A.; Fasoulakis, Z.; Prokopakis, I.; Varthaliti, A.; Karasmani, C.; Ntounis, T.; Domali, E.; Theodora, M.; et al. Maternal Infection and Preterm Birth: From Molecular Basis to Clinical Implications. Children 2023, 10, 907. [Google Scholar] [CrossRef]
- Turunen, J.; Tejesvi, M.V.; Suokas, M.; Virtanen, N.; Paalanne, N.; Kaisanlahti, A.; Reunanen, J.; Tapiainen, T. Bacterial extracellular vesicles in the microbiome of first-pass meconium in newborn infants. Pediatr. Res. 2023, 93, 887–896. [Google Scholar] [CrossRef]
- Kaisanlahti, A.; Turunen, J.; Byts, N.; Samoylenko, A.; Bart, G.; Virtanen, N.; Tejesvi, M.V.; Zhyvolozhnyi, A.; Sarfraz, S.; Kumpula, S.; et al. Maternal microbiota communicates with the fetus through microbiota-derived extracellular vesicles. Microbiome 2023, 11, 249. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Wang, J. Maternal and infant microbiome: Next-generation indicators and targets for intergenerational health and nutrition care. Protein Cell 2023, 14, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xiao, X. The role of gut microbiota in the effects of maternal obesity during pregnancy on offspring metabolism. Biosci. Rep. 2018, 38, BSR20171234. [Google Scholar] [CrossRef]
- Kielenniva, K.; Ainonen, S.; Vanni, P.; Paalanne, N.; Renko, M.; Salo, J.; Tejesvi, M.V.; Pokka, T.; Pirttila, A.M.; Tapiainen, T. Microbiota of the first-pass meconium and subsequent atopic and allergic disorders in children. Clin. Exp. Allergy 2022, 52, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Korpela, K.; Salonen, A.; Saxen, H.; Nikkonen, A.; Peltola, V.; Jaakkola, T.; de Vos, W.; Kolho, K.L. Antibiotics in early life associate with specific gut microbiota signatures in a prospective longitudinal infant cohort. Pediatr. Res. 2020, 88, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Nomura, Y.; Bashir, A.; Fernandez-Hernandez, H.; Itzkowitz, S.; Pei, Z.; Stone, J.; Loudon, H.; Peter, I. Diversified microbiota of meconium is affected by maternal diabetes status. PLoS ONE 2013, 8, e78257. [Google Scholar] [CrossRef]
- Su, M.; Nie, Y.; Shao, R.; Duan, S.; Jiang, Y.; Wang, M.; Xing, Z.; Sun, Q.; Liu, X.; Xu, W. Diversified gut microbiota in newborns of mothers with gestational diabetes mellitus. PLoS ONE 2018, 13, e0205695. [Google Scholar] [CrossRef]
- Chu, D.M.; Antony, K.M.; Ma, J.; Prince, A.L.; Showalter, L.; Moller, M.; Aagaard, K.M. The early infant gut microbiome varies in association with a maternal high-fat diet. Genome Med. 2016, 8, 77. [Google Scholar] [CrossRef]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6, 23129. [Google Scholar] [CrossRef]
- He, Q.; Kwok, L.Y.; Xi, X.; Zhong, Z.; Ma, T.; Xu, H.; Meng, H.; Zhao, F.; Zhang, H. The meconium microbiota shares more features with the amniotic fluid microbiota than the maternal fecal and vaginal microbiota. Gut Microbes 2020, 12, 1794266. [Google Scholar] [CrossRef]
- Miller, C.B.; Benny, P.; Riel, J.; Boushey, C.; Perez, R.; Khadka, V.; Qin, Y.; Maunakea, A.K.; Lee, M.J. Adherence to Mediterranean diet impacts gastrointestinal microbial diversity throughout pregnancy. BMC Pregnancy Childbirth 2021, 21, 558. [Google Scholar] [CrossRef]
- Institute of Medicine and International Pregnancy Weight Gain; National Research Council Committee to Reexamine. The National Academies Collection: Reports funded by National Institutes of Health. In Weight Gain during Pregnancy: Reexamining the Guidelines; Rasmussen, K.M., Yaktine, A.L., Eds.; The National Academies Collection: Reports funded by National Institutes of Health; National Academies Press: Washington, DC, USA, 2009. [Google Scholar]
- Barb, J.J.; Oler, A.J.; Kim, H.S.; Chalmers, N.; Wallen, G.R.; Cashion, A.; Munson, P.J.; Ames, N.J. Development of an Analysis Pipeline Characterizing Multiple Hypervariable Regions of 16S rRNA Using Mock Samples. PLoS ONE 2016, 11, e0148047. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Liang, X.; Niu, Z.Y.; Jin, Q.; Zeng, X.Q.; Wang, W.X.; Li, M.Y.; Chen, X.R.; Meng, H.Y.; Shen, R.; et al. Is the delivery mode a critical factor for the microbial communities in the meconium? EBioMedicine 2019, 49, 354–363. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, J.; Shi, W.; Du, N.; Xu, X.; Zhang, Y.; Ji, P.; Zhang, F.; Jia, Z.; Wang, Y.; et al. Dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus. Gut 2018, 67, 1614–1625. [Google Scholar] [CrossRef] [PubMed]
- Korpela, K.; Renko, M.; Paalanne, N.; Vänni, P.; Salo, J.; Tejesvi, M.; Koivusaari, P.; Pokka, T.; Kaukola, T.; Pirttilä, A.M.; et al. Microbiome of the first stool after birth and infantile colic. Pediatr. Res. 2020, 88, 776–783. [Google Scholar] [CrossRef]
- Graf, J.; Ledala, N.; Caimano, M.J.; Jackson, E.; Gratalo, D.; Fasulo, D.; Driscoll, M.D.; Coleman, S.; Matson, A.P. High-Resolution Differentiation of Enteric Bacteria in Premature Infant Fecal Microbiomes Using a Novel rRNA Amplicon. mBio 2021, 12, e03656-20. [Google Scholar] [CrossRef] [PubMed]
- Wilczyńska, P.; Skarżyńska, E.; Lisowska-Myjak, B. Meconium microbiome as a new source of information about long-term health and disease: Questions and answers. J. Matern.-Fetal Neonatal Med. 2019, 32, 681–686. [Google Scholar] [CrossRef]
- Younge, N.; McCann, J.R.; Ballard, J.; Plunkett, C.; Akhtar, S.; Araújo-Pérez, F.; Murtha, A.; Brandon, D.; Seed, P.C. Fetal exposure to the maternal microbiota in humans and mice. JCI Insight 2019, 4, e127806. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline Demographics of Participants with Matched Neonatal Meconium Samples n = 21 | |
|---|---|
| Maternal Age (years), Median [SD] | 28.6 [5.4] |
| Maternal Ethnicity | |
| Filipino | 5 |
| Japanese | 6 |
| Native Hawaiian | 4 |
| Non-Hispanic White | 6 |
| Parity | |
| Nulliparous | 8 |
| Primiparous | 10 |
| Multiparous | 3 |
| Mode of Delivery | |
| Vaginal | 17 |
| Cesarean Delivery | 4 |
| Maternal Obesity | 4/21 (19%) |
| Excess Gestational Weight Gain | 5/21 (23.8%) |
| Pregnancy Complications | |
| Gestational Diabetes | 2 |
| Pregnancy Associated Hypertension | 4 |
| Preterm Birth | 1 |
| Neonatal Birth Weight (grams), Mean [SD] | 3203.62 [561.7] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, C.; Luu, K.; Mikami, B.; Riel, J.; Qin, Y.; Khadka, V.; Lee, M.-J. Temporal Investigation of the Maternal Origins of Fetal Gut Microbiota. Microorganisms 2024, 12, 1865. https://doi.org/10.3390/microorganisms12091865
Miller C, Luu K, Mikami B, Riel J, Qin Y, Khadka V, Lee M-J. Temporal Investigation of the Maternal Origins of Fetal Gut Microbiota. Microorganisms. 2024; 12(9):1865. https://doi.org/10.3390/microorganisms12091865
Chicago/Turabian StyleMiller, Corrie, Kayti Luu, Brandi Mikami, Jonathan Riel, Yujia Qin, Vedbar Khadka, and Men-Jean Lee. 2024. "Temporal Investigation of the Maternal Origins of Fetal Gut Microbiota" Microorganisms 12, no. 9: 1865. https://doi.org/10.3390/microorganisms12091865