
Intergenerational Transmission of Gut Microbiome from Infected and Non-Infected Salmonella pullorum Hens


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal Experiment and Sample Collection
2.3. 16S rRNA Sequencing and Bioinformatics Analysis
2.4. Statistical Analysis
3. Results
3.1. DNA Sequence Data of Samples Among Different Treatments
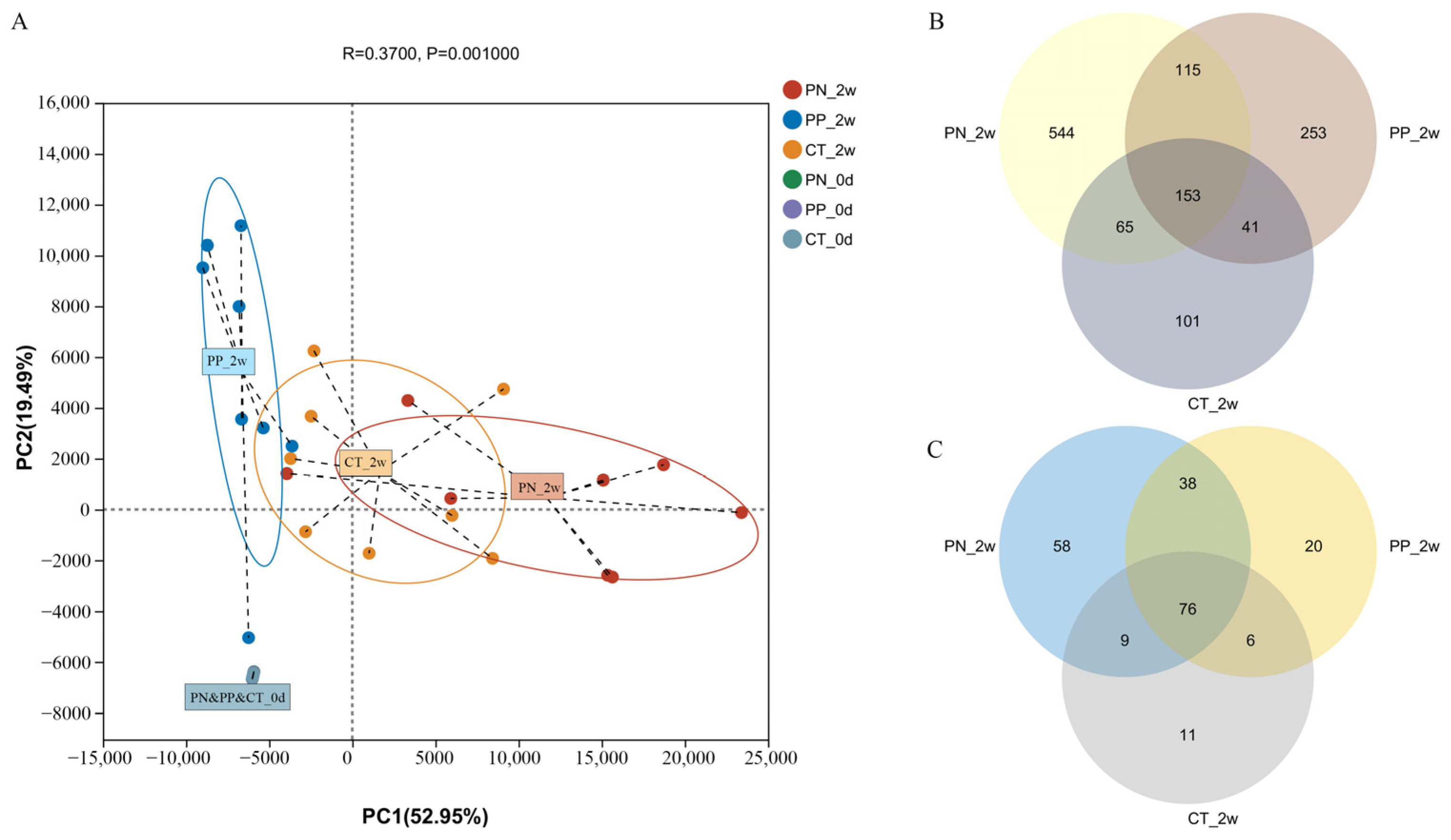
Differences in Reconstituting the Gut Microbiota Structure Between S. pullorum-Negative Transplantation Chicks and S. pullorum-Positive Transplantation Chicks
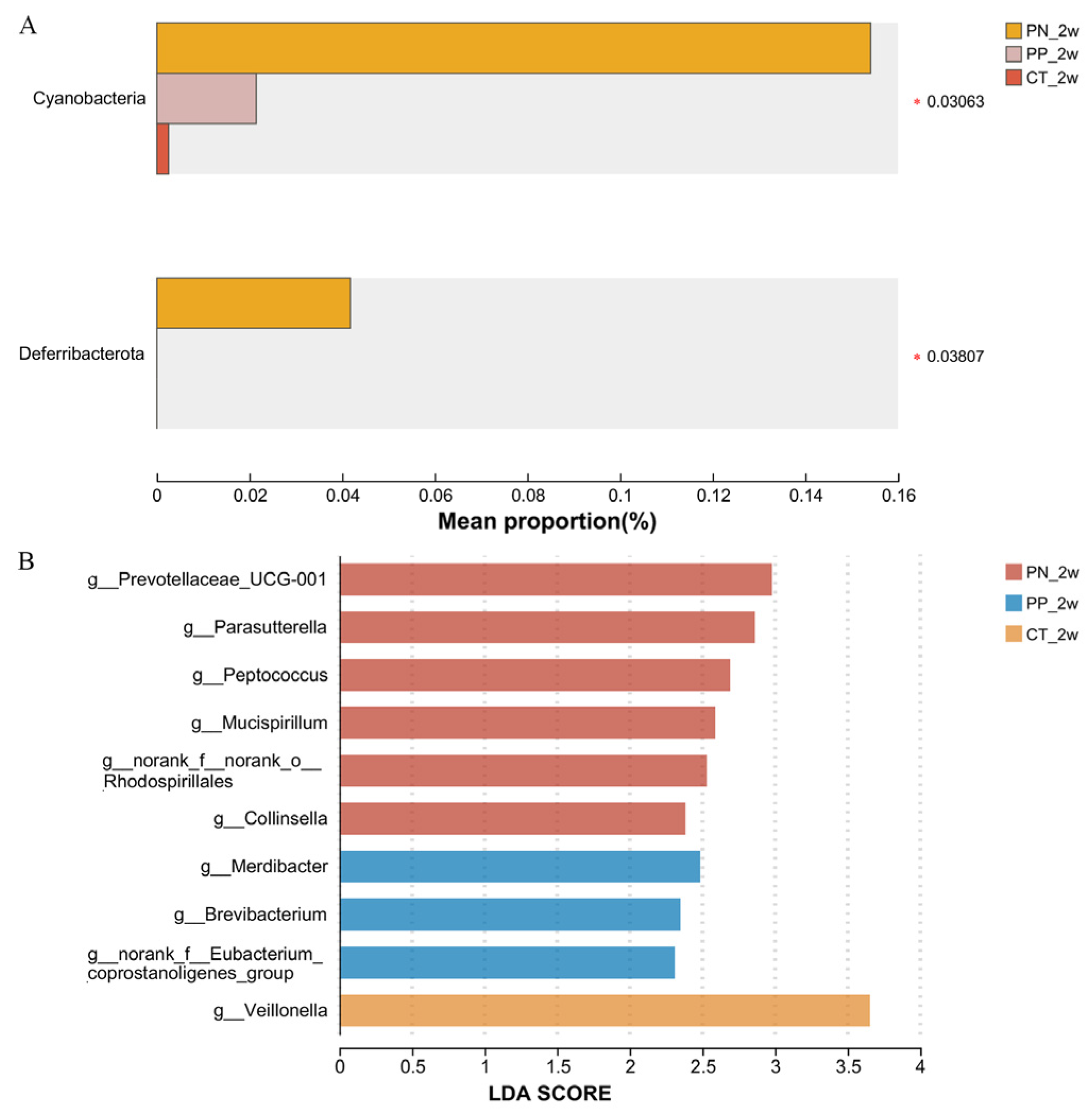
3.2. Identification of Pullorum Disease-Associated Bacterial Taxa Associated with Pullorum Disease That Can Be Transmitted Across Generations
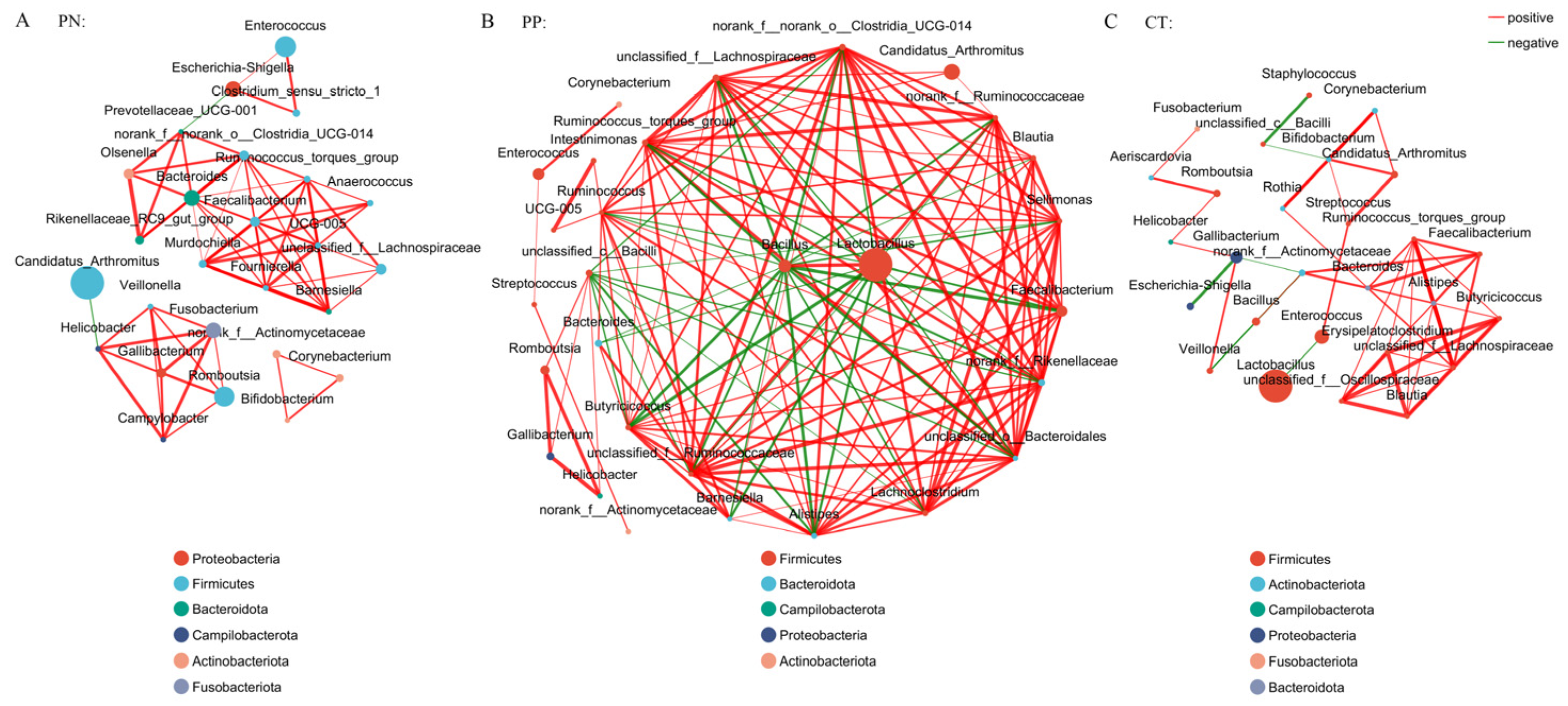
3.3. Analysis of the Co-Occurrence Network of Microorganisms Between S. pullorum-Negative Hens and S. pullorum-Positive Hens
3.4. Prediction Functions of Microbial Metabolism in the Chicks with Different Treatments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FMT | Fecal microbiota transplantation |
| CT | Control team |
| PN | S. pullorum-negative transplantation team |
| PP | S. pullorum-positive transplantation team |
| PD | Pullorum disease |
| S. pullorum | Salmonella pullorum |
| NCBI | National center for biotechnology information |
| SRA | Sequence read archive |
| ASVs | Amplicon sequence variants |
| PCA | Principal component analysis |
| PC1 | Principal component 1 |
| ANOSIM | Analysis of similarities |
| LDA | Linear discriminant analysis |
| LEfSe | Linear discriminant analysis effect size |
| PICRUSt | Phylogenetic investigation of communities by reconstruction of unobserved states |
References
- Li, X.; Nie, C.; Zhang, Z.; Wang, Q.; Shao, P.; Zhao, Q.; Chen, Y.; Wang, D.; Li, Y.; Jiao, W.; et al. Evaluation of genetic resistance to Salmonella Pullorum in three chicken lines. Poult. Sci. 2018, 97, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Li, T.; Liu, F.; Cheng, Y.; Guo, X.; Wen, G.; Luo, Q.; Shao, H.; Pan, Z.; et al. Characterization of Salmonella spp. isolated from chickens in Central China. BMC Vet Res. 2020, 16, 299. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Kang, X.; Zhou, K.; Yue, M. A global dataset for prevalence of Salmonella Gallinarum between 1945 and 2021. Sci. Data 2022, 9, 495. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Ran, X.; Qiu, H.; Zhao, S.; Hu, Z.; Wang, J.; Ni, H.; Wen, X. Seroprevalence of pullorum disease in chicken across mainland China from 1982 to 2020: A systematic review and meta-analysis. Res. Vet Sci. 2022, 152, 156–166. [Google Scholar] [CrossRef]
- Jiang, Z.; Kang, X.; Song, Y.; Zhou, X.; Yue, M. Identification and Evaluation of Novel Antigen Candidates against Salmonella Pullorum Infection Using Reverse Vaccinology. Vaccines 2023, 11, 865. [Google Scholar] [CrossRef]
- Hazards, E.P.o.B.; Koutsoumanis, K.; Allende, A.; Alvarez-Ordonez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; De Cesare, A.; Herman, L.; Hilbert, F.; et al. Salmonella control in poultry flocks and its public health impact. EFSA J. 2019, 17, e05596. [Google Scholar] [CrossRef]
- Barrow, P.A.; Jones, M.A.; Smith, A.L.; Wigley, P. The long view: Salmonella—The last forty years. Avian Pathol. 2012, 41, 413–420. [Google Scholar] [CrossRef]
- Barrow, P.A.; Freitas Neto, O.C. Pullorum disease and fowl typhoid—New thoughts on old diseases: A review. Avian Pathol. 2011, 40, 1–13. [Google Scholar] [CrossRef]
- Khoruts, A.; Sadowsky, M.J. Understanding the mechanisms of faecal microbiota transplantation. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 508–516. [Google Scholar] [CrossRef]
- Britton, R.A.; Young, V.B. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology 2014, 146, 1547–1553. [Google Scholar] [CrossRef]
- Bakken, J.S.; Borody, T.; Brandt, L.J.; Brill, J.V.; Demarco, D.C.; Franzos, M.A.; Kelly, C.; Khoruts, A.; Louie, T.; Martinelli, L.P.; et al. Treating Clostridium difficile infection with fecal microbiota transplantation. Clin. Gastroenterol. Hepatol. 2011, 9, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, I. Composition and Function of Chicken Gut Microbiota. Animals 2020, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Stanley, D.; Hughes, R.J.; Moore, R.J. Microbiota of the chicken gastrointestinal tract: Influence on health, productivity and disease. Appl. Microbiol. Biotechnol. 2014, 98, 4301–4310. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Niu, Q.; Wang, X.; Qi, X.; Cao, C.; Yang, K.; Gu, C.; Zhou, Z.; Huang, Q. Identification of the gut microbiota affecting Salmonella pullorum and their relationship with reproductive performance in hens. Front. Microbiol. 2023, 14, 1216542. [Google Scholar] [CrossRef]
- Li, N.; Zuo, B.; Huang, S.; Zeng, B.; Han, D.; Li, T.; Liu, T.; Wu, Z.; Wei, H.; Zhao, J.; et al. Spatial heterogeneity of bacterial colonization across different gut segments following inter-species microbiota transplantation. Microbiome 2020, 8, 161. [Google Scholar] [CrossRef]
- Janssen, A.W.; Kersten, S. The role of the gut microbiota in metabolic health. FASEB J. 2015, 29, 3111–3123. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudie, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Barberan, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Shivaprasad, H.L. Fowl typhoid and pullorum disease. Rev. Sci. Tech. 2000, 19, 405–424. [Google Scholar] [CrossRef]
- Wigley, P.; Berchieri, A., Jr.; Page, K.L.; Smith, A.L.; Barrow, P.A. Salmonella enterica serovar Pullorum persists in splenic macrophages and in the reproductive tract during persistent, disease-free carriage in chickens. Infect. Immun. 2001, 69, 7873–7879. [Google Scholar] [CrossRef]
- Allegretti, J.R.; Mullish, B.H.; Kelly, C.; Fischer, M. The evolution of the use of faecal microbiota transplantation and emerging therapeutic indications. Lancet 2019, 394, 420–431. [Google Scholar] [CrossRef]
- Zhu, J.; Ding, J.; Yang, K.; Zhou, H.; Yang, W.; Qin, C.; Wang, L.; Xiao, F.; Zhang, B.; Niu, Q.; et al. Microbiome and Microbial Pure Culture Study Reveal Commensal Microorganisms Alleviate Salmonella enterica Serovar Pullorum Infection in Chickens. Microorganisms 2024, 12, 1743. [Google Scholar] [CrossRef]
- Ding, J.; Zhou, H.; Luo, L.; Xiao, L.; Yang, K.; Yang, L.; Zheng, Y.; Xu, K.; He, C.; Han, C.; et al. Heritable Gut Microbiome Associated with Salmonella enterica Serovar Pullorum Infection in Chickens. mSystems 2021, 6, e01192-20. [Google Scholar] [CrossRef]
- Lopez, C.A.; Winter, S.E.; Rivera-Chavez, F.; Xavier, M.N.; Poon, V.; Nuccio, S.P.; Tsolis, R.M.; Baumler, A.J. Phage-mediated acquisition of a type III secreted effector protein boosts growth of Salmonella by nitrate respiration. mBio 2012, 3, e00143-12. [Google Scholar] [CrossRef]
- Lopez, C.A.; Rivera-Chavez, F.; Byndloss, M.X.; Baumler, A.J. The Periplasmic Nitrate Reductase NapABC Supports Luminal Growth of Salmonella enterica Serovar Typhimurium during Colitis. Infect. Immun. 2015, 83, 3470–3478. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, M.; Fang, S.; Huang, X.; He, M.; Ke, S.; Gao, J.; Wu, J.; Zhou, Y.; Fu, H.; et al. Evaluating the profound effect of gut microbiome on host appetite in pigs. BMC Microbiol. 2018, 18, 215. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Fang, S.; Wei, H.; He, M.; Fu, H.; Xiong, X.; Zhou, Y.; Wu, J.; Gao, J.; Yang, H.; et al. Prevotella copri increases fat accumulation in pigs fed with formula diets. Microbiome 2021, 9, 175. [Google Scholar] [CrossRef]
- Tett, A.; Pasolli, E.; Masetti, G.; Ercolini, D.; Segata, N. Prevotella diversity, niches and interactions with the human host. Nat Rev. Microbiol. 2021, 19, 585–599. [Google Scholar] [CrossRef]
- Ju, T.; Kong, J.Y.; Stothard, P.; Willing, B.P. Defining the role of Parasutterella, a previously uncharacterized member of the core gut microbiota. ISME J. 2019, 13, 1520–1534. [Google Scholar] [CrossRef]
- Herp, S.; Durai Raj, A.C.; Salvado Silva, M.; Woelfel, S.; Stecher, B. The human symbiont Mucispirillum schaedleri: Causality in health and disease. Med. Microbiol. Immunol. 2021, 210, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Kalinkovich, A.; Livshits, G. A cross talk between dysbiosis and gut-associated immune system governs the development of inflammatory arthropathies. Semin. Arthritis Rheum. 2019, 49, 474–484. [Google Scholar] [CrossRef]
- Balakrishnan, B.; Luckey, D.; Taneja, V. Autoimmunity-Associated Gut Commensals Modulate Gut Permeability and Immunity in Humanized Mice. Mil. Med. 2019, 184, 529–536. [Google Scholar] [CrossRef]
- Hirayama, M.; Nishiwaki, H.; Hamaguchi, T.; Ito, M.; Ueyama, J.; Maeda, T.; Kashihara, K.; Tsuboi, Y.; Ohno, K. Intestinal Collinsella may mitigate infection and exacerbation of COVID-19 by producing ursodeoxycholate. PLoS ONE 2021, 16, e0260451. [Google Scholar] [CrossRef]
- Chen, C.; Li, J.; Zhang, H.; Xie, Y.; Xiong, L.; Liu, H.; Wang, F. Effects of a probiotic on the growth performance, intestinal flora, and immune function of chicks infected with Salmonella pullorum. Poult. Sci. 2020, 99, 5316–5323. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, Q.; Yang, K.; Zhou, Z.; Huang, Q.; Wang, J. Intergenerational Transmission of Gut Microbiome from Infected and Non-Infected Salmonella pullorum Hens. Microorganisms 2025, 13, 640. https://doi.org/10.3390/microorganisms13030640
Niu Q, Yang K, Zhou Z, Huang Q, Wang J. Intergenerational Transmission of Gut Microbiome from Infected and Non-Infected Salmonella pullorum Hens. Microorganisms. 2025; 13(3):640. https://doi.org/10.3390/microorganisms13030640
Chicago/Turabian StyleNiu, Qing, Kaixuan Yang, Zhenxiang Zhou, Qizhong Huang, and Junliang Wang. 2025. "Intergenerational Transmission of Gut Microbiome from Infected and Non-Infected Salmonella pullorum Hens" Microorganisms 13, no. 3: 640. https://doi.org/10.3390/microorganisms13030640
APA StyleNiu, Q., Yang, K., Zhou, Z., Huang, Q., & Wang, J. (2025). Intergenerational Transmission of Gut Microbiome from Infected and Non-Infected Salmonella pullorum Hens. Microorganisms, 13(3), 640. https://doi.org/10.3390/microorganisms13030640