
Safety and Efficacy of Ephedrine Alkaloids-Free Ephedra Herb Extract (EFE) for Mild COVID-19: A Double-Blind, Placebo-Controlled, Randomized Comparative Trial
 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Patients
2.3. Study Drugs
2.4. Placebo
2.5. Treatment
2.5.1. Part 1
2.5.2. Part 2
2.6. Outcomes
2.7. Statistical Analysis
2.7.1. Part 1
2.7.2. Part 2
2.8. Ethical Considerations
3. Results
3.1. Part 1
3.2. Part 2
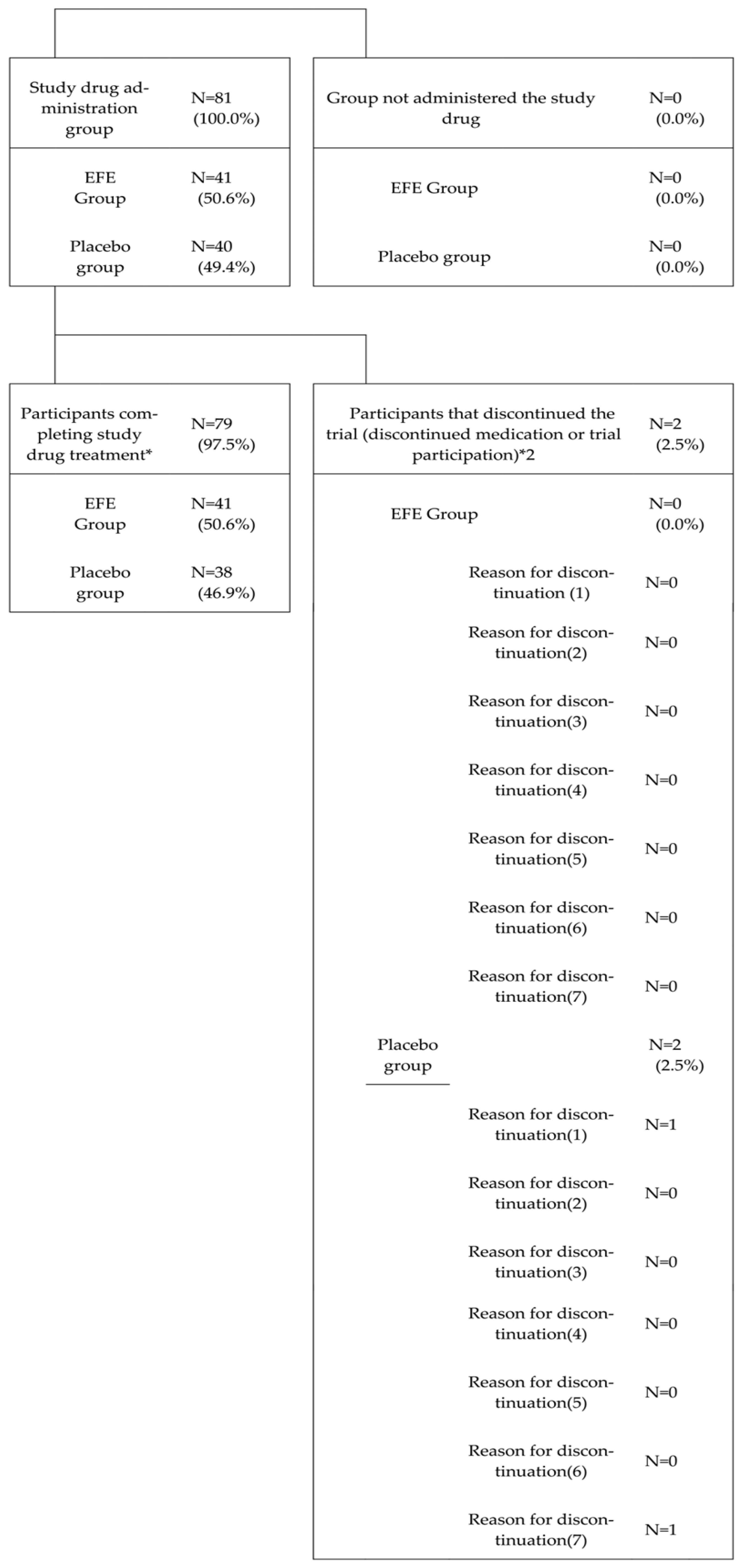
3.2.1. Participant Composition
3.2.2. Patient Background and Physical Findings
3.2.3. Analysis Population
3.2.4. Efficacy
3.2.5. Safety
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thakur, B.; Dubey, P.; Benitez, J.; Torres, J.P.; Reddy, S.; Shokar, N.; Aung, K.; Mukherjee, D.; Dwivedi, A.K. A systematic review and meta-analysis of geographic differences in comorbidities and associated severity and mortality among individuals with COVID-19. Sci. Rep. 2021, 11, 8562. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Peng, F.; Xu, B.; Zhao, J.; Liu, H.; Peng, J.; Li, Q.; Jiang, C.; Zhou, Y.; Liu, S.; et al. Risk factors of critical & mortal COVID-19 cases: A systematic literature review and meta-analysis. J. Infect. 2020, 81, e16–e25. [Google Scholar] [PubMed]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura, T.; Sonoyama, T.; Ichihashi, G.; Sanaki, T.; Tsuge, Y.; Uehara, T.; Mukae, H. A phase 2/3 study of S-217622 in participants with SARS-CoV-2 infection (Phase 3 part). Medicine 2023, 102, e33024. [Google Scholar] [CrossRef]
- Agrawal, U.; Bedston, S.; McCowan, C.; Oke, J.; Patterson, L.; Robertson, C.; Akbari, A.; Azcoaga-Lorenzo, A.; Bradley, D.T.; Fagbamigbe, A.F.; et al. Severe COVID-19 outcomes after full vaccination of primary schedule and initial boosters: Pooled analysis of national prospective cohort studies of 30 million individuals in England, Northern Ireland, Scotland, and Wales. Lancet 2022, 400, 1305–1320. [Google Scholar] [CrossRef]
- Heilmann, E.; Costacurta, F.; Moghadasi, S.A.; Ye, C.; Pavan, M.; Bassani, D.; Volland, A.; Ascher, C.; Weiss, A.K.H.; Bante, D.; et al. SARS-CoV-2 3CL(pro) mutations selected in a VSV-based system confer resistance to nirmatrelvir, ensitrelvir, and GC376. Sci. Transl. Med. 2023, 15, eabq7360. [Google Scholar] [CrossRef]
- Hogan, J.I.; Duerr, R.; Dimartino, D.; Marier, C.; Hochman, S.E.; Mehta, S.; Wang, G.; Heguy, A. Remdesivir Resistance in Transplant Recipients with Persistent Coronavirus Disease 2019. Clin. Infect. Dis. 2023, 76, 342–345. [Google Scholar] [CrossRef]
- Hori, S.; Mihaylov, I.; Vasconcelos, J.C.; McCoubrie, M. Patterns of complementary and alternative medicine use amongst outpatients in Tokyo, Japan. BMC Complement. Altern. Med. 2008, 8, 14. [Google Scholar] [CrossRef]
- Terasawa, K. Evidence-based Reconstruction of Kampo Medicine: Part I-Is Kampo CAM? Evid.-Based Complement. Altern. Med. 2004, 1, 11–16. [Google Scholar] [CrossRef]
- Moschik, E.C.; Mercado, C.; Yoshino, T.; Matsuura, K.; Watanabe, K. Usage and attitudes of physicians in Japan concerning traditional Japanese medicine (kampo medicine): A descriptive evaluation of a representative questionnaire-based survey. Evid.-Based Complement. Altern. Med. 2012, 2012, 139818. [Google Scholar] [CrossRef]
- Nabeshima, S.; Kashiwagi, K.; Ajisaka, K.; Masui, S.; Takeoka, H.; Ikematsu, H.; Kashiwagi, S. A randomized, controlled trial comparing traditional herbal medicine and neuraminidase inhibitors in the treatment of seasonal influenza. J. Infect. Chemother. 2012, 18, 534–543. [Google Scholar] [CrossRef]
- Toriumi, Y.; Kamei, T.; Murata, K.; Takahashi, I.; Suzuki, N.; Mazda, O. Utility of Maoto in an influenza season where reduced effectiveness of oseltamivir was observed—A clinical, non-randomized study in children. Forsch. Komplementmed. 2012, 19, 179–186. [Google Scholar] [CrossRef]
- Mantani, N.; Andoh, T.; Kawamata, H.; Terasawa, K.; Ochiai, H. Inhibitory effect of Ephedrae herba, an oriental traditional medicine, on the growth of influenza A/PR/8 virus in MDCK cells. Antivir. Res. 1999, 44, 193–200. [Google Scholar] [CrossRef]
- Odaguchi, H.; Hyuga, S.; Sekine, M.; Nakamori, S.; Takemoto, H.; Huang, X.; Oshima, N.; Shimada, N.; Yang, J.; Amakura, Y.; et al. The Adverse Effects of Ephedra Herb and the Safety of Ephedrine Alkaloids-free Ephedra Herb Extract (EFE). Yakugaku Zasshi 2019, 139, 1417–1425. [Google Scholar] [CrossRef]
- Oshima, N.; Yamashita, T.; Hyuga, S.; Hyuga, M.; Kamakura, H.; Yoshimura, M.; Maruyama, T.; Hakamatsuka, T.; Amakura, Y.; Hanawa, T.; et al. Efficiently prepared ephedrine alkaloids-free Ephedra Herb extract: A putative marker and antiproliferative effects. J. Nat. Med. 2016, 70, 554–562. [Google Scholar] [CrossRef]
- Hyuga, S.; Hyuga, M.; Oshima, N.; Maruyama, T.; Kamakura, H.; Yamashita, T.; Yoshimura, M.; Amakura, Y.; Hakamatsuka, T.; Odaguchi, H.; et al. Ephedrine alkaloids-free Ephedra Herb extract: A safer alternative to ephedra with comparable analgesic, anticancer, and anti-influenza activities. J. Nat. Med. 2016, 70, 571–583. [Google Scholar] [CrossRef]
- Nakamori, S.; Takahashi, J.; Hyuga, S.; Yang, J.; Takemoto, H.; Maruyama, T.; Oshima, N.; Uchiyama, N.; Amakura, Y.; Hyuga, M.; et al. Analgesic Effects of Ephedra Herb Extract, Ephedrine Alkaloids-Free Ephedra Herb Extract, Ephedrine, and Pseudoephedrine on Formalin-Induced Pain. Biol. Pharm. Bull. 2019, 42, 1538–1544. [Google Scholar] [CrossRef]
- Takemoto, H.; Takahashi, J.; Hyuga, S.; Odaguchi, H.; Uchiyama, N.; Maruyama, T.; Yamashita, T.; Hyuga, M.; Oshima, N.; Amakura, Y.; et al. Ephedrine Alkaloids-Free Ephedra Herb Extract, EFE, Has No Adverse Effects Such as Excitation, Insomnia, and Arrhythmias. Biol. Pharm. Bull. 2018, 41, 247–253. [Google Scholar] [CrossRef]
- Odaguchi, H.; Sekine, M.; Hyuga, S.; Hanawa, T.; Hoshi, K.; Sasaki, Y.; Aso, M.; Yang, J.; Hyuga, M.; Kobayashi, Y.; et al. A Double-Blind, Randomized, Crossover Comparative Study for Evaluating the Clinical Safety of Ephedrine Alkaloids-Free Ephedra Herb Extract (EFE). Evid.-Based Complement. Altern. Med. 2018, 2018, 4625358. [Google Scholar] [CrossRef]
- Uema, M.; Hyuga, M.; Yonemitsu, K.; Hyuga, S.; Amakura, Y.; Uchiyama, N.; Mizoguchi, K.; Odaguchi, H.; Goda, Y. Antiviral Effect of Ephedrine Alkaloids-Free Ephedra Herb Extract against SARS-CoV-2 In Vitro. Microorganisms 2023, 11, 534. [Google Scholar] [CrossRef]
- Nabeshima, A.; Sakamoto, A.; Iwata, K.; Kitamura, Y.; Masui, S.; Inomata, S.; Iida, M.; Iida, T.; Nabeshima, S. Maoto, a traditional herbal medicine, for post-exposure prophylaxis for Japanese healthcare workers exposed to COVID-19: A single center study. J. Infect. Chemother. 2022, 28, 907–911. [Google Scholar] [CrossRef]
- Takayama, S.; Namiki, T.; Odaguchi, H.; Arita, R.; Hisanaga, A.; Mitani, K.; Ito, T. Prevention and Recovery of COVID-19 Patients with Kampo Medicine: Review of Case Reports and Ongoing Clinical Trials. Front. Pharmacol. 2021, 12, 656246. [Google Scholar] [CrossRef]
- Dousari, A.S.; Amroabadi, M.K.; Neyestani, Z.S.; Moghadam, M.T.; Satarzadeh, N. The use of ephedra herbs in the treatment of COVID-19. Avicenna J. Phytomed. 2023, 13, 231–239. [Google Scholar]
- Yoshimura, M.; Amakura, Y.; Hyuga, S.; Hyuga, M.; Nakamori, S.; Maruyama, T.; Oshima, N.; Uchiyama, N.; Yang, J.; Oka, H.; et al. Quality evaluation and characterization of fractions with biological activity from Ephedra herb Extract and ephedrine alkaloids-free Ephedra Herb extract. Chem. Pharm. Bull. 2020, 68, 140–149. [Google Scholar] [CrossRef]
- Arabi, M.; Al-Najjar, Y.; Mhaimeed, N.; Salameh, M.A.; Paul, P.; AlAnni, J.; Abdelati, A.A.; Laswi, I.; Khanjar, B.; Al-Ali, D.; et al. Severity of the Omicron SARS-CoV-2 variant compared with the previous lineages: A systematic review. J. Cell Mol. Med. 2023, 27, 1443–1464. [Google Scholar] [CrossRef]
- Nyberg, T.; Ferguson, N.M.; Nash, S.G.; Webster, H.H.; Flaxman, S.; Andrews, N.; Hinsley, W.; Bernal, J.L.; Kall, M.; Bhatt, S.; et al. Comparative analysis of the risks of hospitalisation and death associated with SARS-CoV-2 Omicron (B.1.1.529) and delta (B.1.617.2) variants in England: A Cohort Study. Lancet 2022, 399, 1303–1312. [Google Scholar] [CrossRef]
- Havers, F.P.; Pham, H.; Taylor, C.A.; Whitaker, M.; Patel, K.; Anglin, O.; Kambhampati, A.K.; Milucky, J.; Zell, E.; Moline, H.L.; et al. COVID-19-Associated Hospitalizations Among Vaccinated and Unvaccinated Adults 18 Years or Older in 13 US States, January 2021 to April 2022. JAMA Intern. Med. 2022, 182, 1071–1081. [Google Scholar] [CrossRef]
- Tenforde, M.W.; Self, W.H.; Gaglani, M.; Ginde, A.A.; Douin, D.J.; Talbot, H.K.; Casey, J.D.; Mohr, N.M.; Zepeski, A.; McNeal, T.; et al. Effectiveness of mRNA Vaccination in Preventing COVID-19-Associated Invasive Mechanical Ventilation and Death—United States, March 2021–January 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 459–465. [Google Scholar] [CrossRef]
- Cheung, K.S.; Hung, I.F.N.; Chan, P.P.Y.; Lung, K.C.; Tso, E.; Liu, R.; Ng, Y.Y.; Chu, M.Y.; Chung, T.W.; Tam, A.R.; et al. Gastrointestinal Manifestations of SARS-CoV-2 Infection and Virus Load in Fecal Samples from a Hong Kong Cohort: Systematic Review and Meta-Analysis. Gastroenterology 2020, 159, 81–95. [Google Scholar] [CrossRef]
- Caldera-Crespo, L.A.; Paidas, M.J.; Roy, S.; Schulman, C.I.; Kenyon, N.S.; Daunert, S.; Jayakumar, A.R. Experimental Models of COVID-19. Front. Cell Infect. Microbiol. 2021, 11, 792584. [Google Scholar] [CrossRef]
- De Albuquerque, N.; Baig, E.; Ma, X.; Zhang, J.; He, W.; Rowe, A.; Habal, M.; Liu, M.; Shalev, I.; Downey, G.P.; et al. Murine hepatitis virus strain 1 produces a clinically relevant model of severe acute respiratory syndrome in A/J mice. J. Virol. 2006, 80, 10382–10394. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| EFE Group (N = 5) | ||
|---|---|---|
| Events | Number of Events | Number of Affected Participants (%) |
| Adverse events | ||
| All events | 6 | 2 (40.0) |
| Mental disorder | 1 | 1 (20.0) |
| Initial insomnia | 1 | 1 (20.0) |
| Gastrointestinal disorder | 2 | 1 (20.0) |
| Constipation | 1 | 1 (20.0) |
| Diarrhea | 1 | 1 (20.0) |
| Clinical laboratory results | 3 | 1 (20.0) |
| C-reactive protein increased | 1 | 1 (20.0) |
| Neutrophil count increased | 1 | 1 (20.0) |
| White blood cell count increased | 1 | 1 (20.0) |
| Side effects | ||
| All events | 3 | 1 (20.0) |
| Clinical laboratory results | 3 | 1 (20.0) |
| C-reactive protein increased | 1 | 1 (20.0) |
| Neutrophil count increased | 1 | 1 (20.0) |
| White blood cell count increased | 1 | 1 (20.0) |
| Analyzed population: mITT | |||||
| Primary analysis | |||||
| Time | Group | Target number of participants | Number of participants with non-aggravation (%) | Two-sided 95% CI | Chi-square test |
| Day 15 | EFE group | 38 | 38 (100.0) | 90.7–100.0 | p = 0.146 |
| Placebo group | 37 | 35 (94.6) | 81.8–99.3 | ||
| Analyzed population: PPS | |||||
| Secondary analysis | |||||
| Time | Group | Target number of participants | Number of participants with non-aggravation (%) | Two-sided 95% CI | Chi-square test |
| Day 15 | EFE group | 33 | 33 (100.0) | 89.4–100.0 | p = 0.306 |
| Placebo group | 32 | 31 (96.9) | 83.8–99.9 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odaguchi, H.; Hyuga, S.; Sekine, M.; Michimae, H.; Hyuga, M.; Uchiyama, N.; Uema, M.; Kumagai, Y.; Suzuki, Y.; Nabeshima, S.; et al. Safety and Efficacy of Ephedrine Alkaloids-Free Ephedra Herb Extract (EFE) for Mild COVID-19: A Double-Blind, Placebo-Controlled, Randomized Comparative Trial. Microorganisms 2025, 13, 641. https://doi.org/10.3390/microorganisms13030641
Odaguchi H, Hyuga S, Sekine M, Michimae H, Hyuga M, Uchiyama N, Uema M, Kumagai Y, Suzuki Y, Nabeshima S, et al. Safety and Efficacy of Ephedrine Alkaloids-Free Ephedra Herb Extract (EFE) for Mild COVID-19: A Double-Blind, Placebo-Controlled, Randomized Comparative Trial. Microorganisms. 2025; 13(3):641. https://doi.org/10.3390/microorganisms13030641
Chicago/Turabian StyleOdaguchi, Hiroshi, Sumiko Hyuga, Mariko Sekine, Hirofumi Michimae, Masashi Hyuga, Nahoko Uchiyama, Masashi Uema, Yuji Kumagai, Yusuke Suzuki, Shigeki Nabeshima, and et al. 2025. "Safety and Efficacy of Ephedrine Alkaloids-Free Ephedra Herb Extract (EFE) for Mild COVID-19: A Double-Blind, Placebo-Controlled, Randomized Comparative Trial" Microorganisms 13, no. 3: 641. https://doi.org/10.3390/microorganisms13030641
APA StyleOdaguchi, H., Hyuga, S., Sekine, M., Michimae, H., Hyuga, M., Uchiyama, N., Uema, M., Kumagai, Y., Suzuki, Y., Nabeshima, S., Omagari, N., Doi, Y., Yamaoka, K., Miyazaki, K., Fuji, S., Umezawa, Y., Kodera, S., Nagashima, H., Hirose, W., & Goda, Y. (2025). Safety and Efficacy of Ephedrine Alkaloids-Free Ephedra Herb Extract (EFE) for Mild COVID-19: A Double-Blind, Placebo-Controlled, Randomized Comparative Trial. Microorganisms, 13(3), 641. https://doi.org/10.3390/microorganisms13030641