

Antibiotic-Resistant Pseudomonas aeruginosa: Current Challenges and Emerging Alternative Therapies

Abstract
:1. Introduction
2. Mechanisms of Antibiotic Resistance in Pseudomonas aeruginosa
2.1. Intrinsic Resistance
2.1.1. Active Transport Mode: Efflux Pumps
2.1.2. Low Membrane Permeability to Antibiotics
2.2. Acquired Resistance
2.2.1. Horizontal Gene Transfer
2.2.2. Mutation-Accumulated Antibiotic Resistance
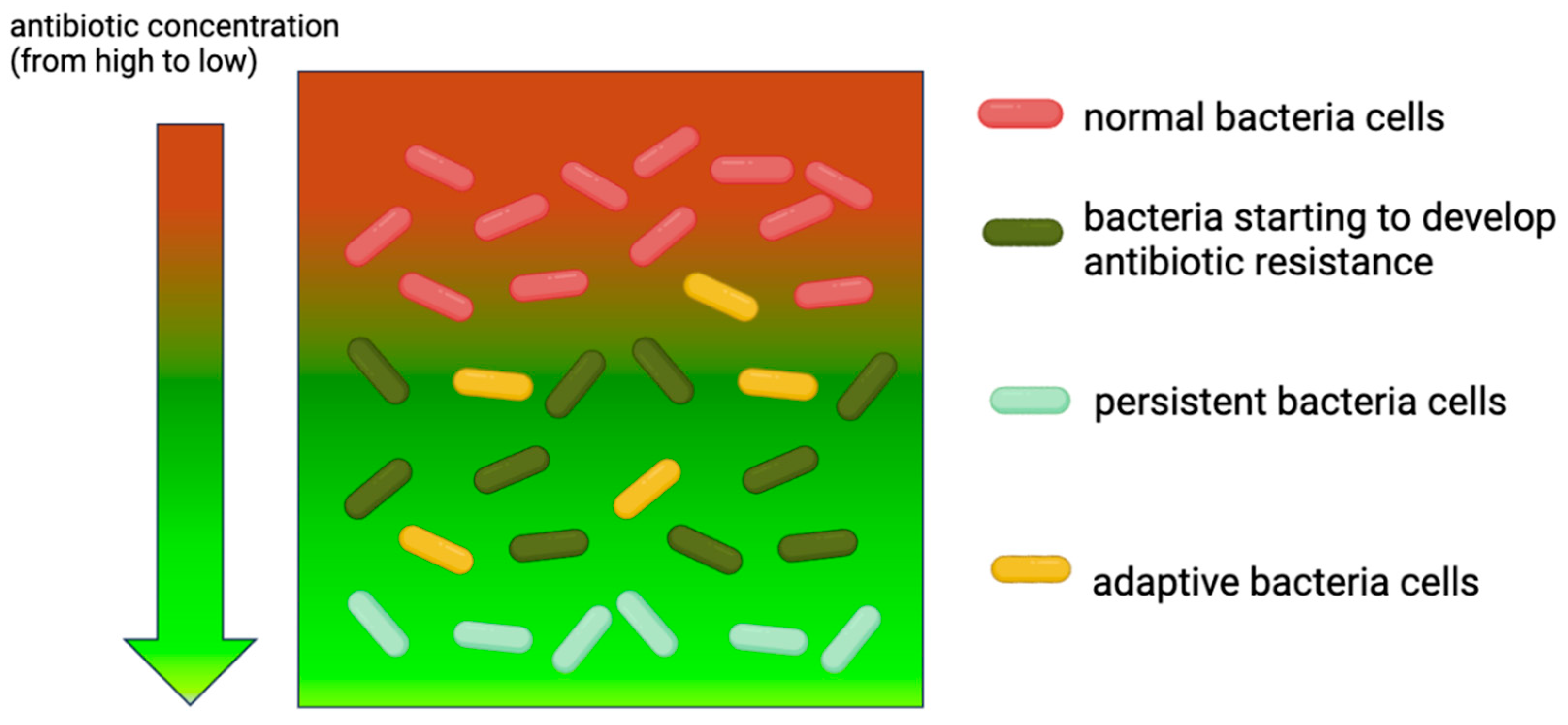
2.3. Adaptive Resistance
2.3.1. Formation of Biofilm
2.3.2. Formation of Persister Cells
2.4. Current First-Line Antibiotics Used to Treat MDR Pseudomonas Infections
3. Current Alternative Therapeutic Approaches
3.1. Phage Therapy
3.1.1. Overview of Bacteriophages Targeting P. aeruginosa
3.1.2. Advantages and Limitations of Phage Therapy
3.1.3. Clinical Trial Status
3.2. Antimicrobial Peptides (AMPs)
3.2.1. Mechanisms of Action Against P. aeruginosa
3.2.2. Challenges in Stability, Delivery, and Toxicity
3.3. Quorum Sensing Inhibitors (QSIs)
3.3.1. Types of QSIs
3.3.2. Challenges in Developing QSIs, Including Specificity and Potential Resistance
3.4. CRISPR-Cas Systems for Targeted Gene Editing
3.4.1. Concept of Using CRISPR to Disrupt Resistance Genes

{kind=link}
{kind=link}
| Bacteria | Gene | Mechanism | Reference |
|---|---|---|---|
| E. coli | Mcr-1 | Insert CRISPR Cas9 to pCas9 plasmid to resensitize bacteria to Colistin | [229] |
| E. faecalis | ermB, tetM | A constitutively expressed CRISPR/Cas9 was designed using pD1 to reduce antibiotic resistance | [230] |
| K. pneumonia | ramR, tetA, mgrB | Use CRISPR Cas9 to modify pSGKP-spe and pBECKP-spe plasmids to make bacteria sensitive to antibiotics (e.g., Colistin) | [226] |
3.4.2. Progress and Challenges in Clinical Application
3.5. Nanoparticles and Drug Delivery Systems
4. Future Directions and Research Needs
4.1. Need for More Robust Clinical Trials for Alternative Therapies
4.2. Potential Regulatory Challenges in Developing and Approving New Treatments
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Araos, R.; D’Agata, E. Pseudomonas aeruginosa and Other Pseudomonas Species. 2019. Available online: https://scholar.googleusercontent.com/scholar?q=cache:zPAFS5s4TcYJ:scholar.google.com/++Pseudomonas+aeruginosa+and+other+Pseudomonas+species.&hl=en&as_sdt=0,5 (accessed on 9 April 2025).
- Diggle, S.P.; Whiteley, M. Microbe Profile: Pseudomonas aeruginosa: Opportunistic pathogen and lab rat. Microbiology 2020, 166, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, D.; Kollef, M. The Epidemiology and Pathogenesis and Treatment of Pseudomonas aeruginosa Infections: An Update. Drugs 2021, 81, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- El Husseini, N.; Carter, J.A.; Lee, V.T. Urinary tract infections and catheter-associated urinary tract infections caused by Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2024, 88, e00066-22. [Google Scholar] [CrossRef] [PubMed]
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A.A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef]
- Campoccia, D.; Montanaro, L.; Speziale, P.; Arciola, C.R. Antibiotic-loaded biomaterials and the risks for the spread of antibiotic resistance following their prophylactic and therapeutic clinical use. Biomaterials 2010, 31, 6363–6377. [Google Scholar] [CrossRef]
- Qin, S.; Xiao, W.; Zhou, C.; Pu, Q.; Deng, X.; Lan, L.; Liang, H.; Song, X.; Wu, M. Pseudomonas aeruginosa: Pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Signal Transduct. Target. Ther. 2022, 7, 199. [Google Scholar] [CrossRef]
- Pachori, P.; Gothalwal, R.; Gandhi, P. Emergence of antibiotic resistance Pseudomonas aeruginosa in intensive care unit; a critical review. Genes Dis. 2019, 6, 109–119. [Google Scholar] [CrossRef]
- Micek, S.T.; Wunderink, R.G.; Kollef, M.H.; Chen, C.; Rello, J.; Chastre, J.; Antonelli, M.; Welte, T.; Clair, B.; Ostermann, H. An international multicenter retrospective study of Pseudomonas aeruginosa nosocomial pneumonia: Impact of multidrug resistance. Crit. Care 2015, 19, 219. [Google Scholar] [CrossRef]
- Catalano, A.; Iacopetta, D.; Ceramella, J.; Scumaci, D.; Giuzio, F.; Saturnino, C.; Aquaro, S.; Rosano, C.; Sinicropi, M.S. Multidrug resistance (MDR): A widespread phenomenon in pharmacological therapies. Molecules 2022, 27, 616. [Google Scholar] [CrossRef]
- Shamsuzzaman, S. Multidrug-resistant, Extensively drug-resistant and Pandrug-resistant bacteria and antimicrobial therapy in combination. Bangladesh J. Med. Microbiol. 2015, 9, 1–2. [Google Scholar] [CrossRef]
- Bubonja-Sonje, M.; Matovina, M.; Skrobonja, I.; Bedenic, B.; Abram, M. Mechanisms of carbapenem resistance in multidrug-resistant clinical isolates of Pseudomonas aeruginosa from a Croatian hospital. Microb. Drug Resist. 2015, 21, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.-I.; Kim, S.-H.; Park, W.B.; Lee, K.-D.; Kim, H.-B.; Kim, E.-C.; Oh, M.-d.; Choe, K.-W. Bloodstream infections caused by antibiotic-resistant gram-negative bacilli: Risk factors for mortality and impact of inappropriate initial antimicrobial therapy on outcome. Antimicrob. Agents Chemother. 2005, 49, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Thaden, J.T.; Park, L.P.; Maskarinec, S.A.; Ruffin, F.; Fowler Jr, V.G.; Van Duin, D. Results from a 13-year prospective cohort study show increased mortality associated with bloodstream infections caused by Pseudomonas aeruginosa compared to other bacteria. Antimicrob. Agents Chemother. 2017, 61, e02671-16. [Google Scholar] [CrossRef] [PubMed]
- Laborda, P.; Hernando-Amado, S.; Martínez, J.L.; Sanz-García, F. Antibiotic Resistance in Pseudomonas. Adv. Exp. Med. Biol. 2022, 1386, 117–143. [Google Scholar] [CrossRef]
- Theuretzbacher, U. Accelerating resistance, inadequate antibacterial drug pipelines and international responses. Int. J. Antimicrob. Agents 2012, 39, 295–299. [Google Scholar] [CrossRef]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Subedi, D.; Vijay, A.K.; Willcox, M. Overview of mechanisms of antibiotic resistance in Pseudomonas aeruginosa: An ocular perspective. Clin. Exp. Optom. 2018, 101, 162–171. [Google Scholar] [CrossRef]
- Du, D.; Wang-Kan, X.; Neuberger, A.; Van Veen, H.W.; Pos, K.M.; Piddock, L.J.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef]
- Black, P.A.; Warren, R.M.; Louw, G.E.; van Helden, P.D.; Victor, T.C.; Kana, B.D. Energy metabolism and drug efflux in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2491–2503. [Google Scholar] [CrossRef]
- Yamasaki, S.; Zwama, M.; Yoneda, T.; Hayashi-Nishino, M.; Nishino, K. Drug resistance and physiological roles of RND multidrug efflux pumps in Salmonella enterica, Escherichia coli and Pseudomonas aeruginosa. Microbiology 2023, 169, 001322. [Google Scholar] [CrossRef]
- Kumawat, M.; Nabi, B.; Daswani, M.; Viquar, I.; Pal, N.; Sharma, P.; Tiwari, S.; Sarma, D.K.; Shubham, S.; Kumar, M. Role of bacterial efflux pump proteins in antibiotic resistance across microbial species. Microb. Pathog. 2023, 181, 106182. [Google Scholar] [CrossRef] [PubMed]
- Wilton, M.; Charron-Mazenod, L.; Moore, R.; Lewenza, S. Extracellular DNA Acidifies Biofilms and Induces Aminoglycoside Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Mine, T.; Morita, Y.; Kataoka, A.; Mizushima, T.; Tsuchiya, T. Expression in Escherichia coli of a new multidrug efflux pump, MexXY, from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1999, 43, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef]
- Moreira, M.A.S.; Desouza, E.C.; Demoraes, C.A. Multidrug efflux systems in Gram-negative bacteria. Braz. J. Microbiol. 2004, 35, 19. [Google Scholar] [CrossRef]
- Poole, K. Multidrug efflux pumps and antimicrobial resistance in Pseudomonas aeruginosa and related organisms. J. Mol. Microbiol. Biotechnol. 2001, 3, 255–264. [Google Scholar]
- Pasqua, M.; Bonaccorsi di Patti, M.C.; Fanelli, G.; Utsumi, R.; Eguchi, Y.; Trirocco, R.; Prosseda, G.; Grossi, M.; Colonna, B. Host—Bacterial Pathogen Communication: The Wily Role of the Multidrug Efflux Pumps of the MFS Family. Front. Mol. Biosci. 2021, 8, 723274. [Google Scholar] [CrossRef]
- Chung, Y.J.; Saier, M.H., Jr. SMR-type multidrug resistance pumps. Curr. Opin. Drug Discov. Devel. 2001, 4, 237–245. [Google Scholar]
- Greene, N.P.; Kaplan, E.; Crow, A.; Koronakis, V. Antibiotic Resistance Mediated by the MacB ABC Transporter Family: A Structural and Functional Perspective. Front. Microbiol. 2018, 9, 950. [Google Scholar] [CrossRef]
- He, G.X.; Thorpe, C.; Walsh, D.; Crow, R.; Chen, H.; Kumar, S.; Varela, M.F. EmmdR, a new member of the MATE family of multidrug transporters, extrudes quinolones from Enterobacter cloacae. Arch. Microbiol. 2011, 193, 759–765. [Google Scholar] [CrossRef]
- Lorusso, A.B.; Carrara, J.A.; Barroso, C.D.N.; Tuon, F.F.; Faoro, H. Role of Efflux Pumps on Antimicrobial Resistance in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2022, 23, 15779. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.H.; Vidaillac, C.; Yam, J.K.H.; Chua, S.L.; Givskov, M.; Yang, L. In Vitro and In Vivo Efficacy of an LpxC Inhibitor, CHIR-090, Alone or Combined with Colistin against Pseudomonas aeruginosa Biofilm. Antimicrob. Agents Chemother. 2017, 61, e02223-16. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Brinkman, F.S. Function of Pseudomonas porins in uptake and efflux. Annu. Rev. Microbiol. 2002, 56, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, E.; Nestorovich, E.M.; Bezrukov, S.M.; Nikaido, H. Pseudomonas aeruginosa porin OprF exists in two different conformations. J. Biol. Chem. 2006, 281, 16220–16229. [Google Scholar] [CrossRef]
- Charpentier, X.; Polard, P.; Claverys, J.P. Induction of competence for genetic transformation by antibiotics: Convergent evolution of stress responses in distant bacterial species lacking SOS? Curr. Opin. Microbiol. 2012, 15, 570–576. [Google Scholar] [CrossRef]
- Mazaheri Nezhad Fard, R.; Barton, M.D.; Heuzenroeder, M.W. Bacteriophage-mediated transduction of antibiotic resistance in Enterococci. Lett. Appl. Microbiol. 2011, 52, 559–564. [Google Scholar] [CrossRef]
- Smillie, C.; Garcillán-Barcia, M.P.; Francia, M.V.; Rocha, E.P.; de la Cruz, F. Mobility of plasmids. Microbiol. Mol. Biol. Rev. 2010, 74, 434–452. [Google Scholar] [CrossRef]
- Breidenstein, E.B.; de la Fuente-Núñez, C.; Hancock, R.E. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426. [Google Scholar] [CrossRef]
- Morales-Espinosa, R.; Soberón-Chávez, G.; Delgado-Sapién, G.; Sandner-Miranda, L.; Méndez, J.L.; González-Valencia, G.; Cravioto, A. Genetic and Phenotypic Characterization of a Pseudomonas aeruginosa Population with High Frequency of Genomic Islands. PLoS ONE 2012, 7, e37459. [Google Scholar] [CrossRef]
- Cavalcanti, F.L.d.S.; Mirones, C.R.; Paucar, E.R.; Montes, L.Á.; Leal-Balbino, T.C.; Morais, M.M.C.d.; Martínez-Martínez, L.; Ocampo-Sosa, A.A. Mutational and acquired carbapenem resistance mechanisms in multidrug resistant Pseudomonas aeruginosa clinical isolates from Recife, Brazil. Memórias Do Inst. Oswaldo Cruz 2015, 110, 1003–1009. [Google Scholar] [CrossRef]
- Di Pilato, V.; Antonelli, A.; Giani, T.; Henrici De Angelis, L.; Rossolini, G.M.; Pollini, S. Identification of a Novel Plasmid Lineage Associated with the Dissemination of Metallo-β-Lactamase Genes Among Pseudomonads. Front. Microbiol. 2019, 10, 1504. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.-J.; Jeong, S.H. Mobile Carbapenemase Genes in Pseudomonas aeruginosa. Front. Microbiol. 2021, 12, 614058. [Google Scholar] [CrossRef] [PubMed]
- Jacoby George, A. AmpC β-Lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, C.; Song, J.; Velkov, T.; Wang, L.; Zhu, Y.; Li, J. Regulating polymyxin resistance in Gram-negative bacteria: Roles of two-component systems PhoPQ and PmrAB. Future Microbiol. 2020, 15, 445–459. [Google Scholar] [CrossRef]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef]
- Varadarajan, A.R.; Allan, R.N.; Valentin, J.D.P.; Castañeda Ocampo, O.E.; Somerville, V.; Pietsch, F.; Buhmann, M.T.; West, J.; Skipp, P.J.; van der Mei, H.C.; et al. An integrated model system to gain mechanistic insights into biofilm-associated antimicrobial resistance in Pseudomonas aeruginosa MPAO1. npj Biofilms Microbiomes 2020, 6, 46. [Google Scholar] [CrossRef]
- Wang, C.; Wang, J.; Mi, Z. Pseudomonas aeruginosa producing VIM-2 metallo-β-lactamases and carrying two aminoglycoside-modifying enzymes in China. J. Hosp. Infect. 2006, 62, 522–524. [Google Scholar] [CrossRef]
- Cayci, Y.T.; Coban, A.; Gunaydin, M. Investigation of plasmid-mediated quinolone resistance in Pseudomonas aeruginosa clinical isolates. Indian J. Med. Microbiol. 2014, 32, 285–289. [Google Scholar] [CrossRef]
- Cazares, A.; Moore, M.P.; Hall, J.P.J.; Wright, L.L.; Grimes, M.; Emond-Rhéault, J.-G.; Pongchaikul, P.; Santanirand, P.; Levesque, R.C.; Fothergill, J.L.; et al. A megaplasmid family driving dissemination of multidrug resistance in Pseudomonas. Nat. Commun. 2020, 11, 1370. [Google Scholar] [CrossRef]
- Hernández-Ramírez, K.C.; Reyes-Gallegos, R.I.; Chávez-Jacobo, V.M.; Díaz-Magaña, A.; Meza-Carmen, V.; Ramírez-Díaz, M.I. A plasmid-encoded mobile genetic element from Pseudomonas aeruginosa that confers heavy metal resistance and virulence. Plasmid 2018, 98, 15–21. [Google Scholar] [CrossRef]
- López-Causapé, C.; Cabot, G.; Del Barrio-Tofiño, E.; Oliver, A. The Versatile Mutational Resistome of Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 685. [Google Scholar] [CrossRef] [PubMed]
- Landry, R.M.; An, D.; Hupp, J.T.; Singh, P.K.; Parsek, M.R. Mucin–Pseudomonas aeruginosa interactions promote biofilm formation and antibiotic resistance. Mol. Microbiol. 2006, 59, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, M.B.; Gardner, P.P.; Lamont, I.L. The PitA protein contributes to colistin susceptibility in Pseudomonas aeruginosa. PLoS ONE 2023, 18, e0292818. [Google Scholar] [CrossRef] [PubMed]
- Tierney, A.R.; Rather, P.N. Roles of two-component regulatory systems in antibiotic resistance. Future Microbiol. 2019, 14, 533–552. [Google Scholar] [CrossRef]
- Ma, Y.; Aung, T.T.; Lakshminarayanan, R.; Chua, S.L. Biofilm formation and virulence potential of carbapenem-resistant Pseudomonas aeruginosa. Lancet Microbe 2023, 4, e489. [Google Scholar] [CrossRef]
- Yano, H.; Hayashi, W.; Kawakami, S.; Aoki, S.; Anzai, E.; Zuo, H.; Kitamura, N.; Hirabayashi, A.; Kajihara, T.; Kayama, S.; et al. Nationwide genome surveillance of carbapenem-resistant Pseudomonas aeruginosa in Japan. Antimicrob. Agents Chemother. 2024, 68, e01669-23. [Google Scholar] [CrossRef]
- Sandoval-Motta, S.; Aldana, M. Adaptive resistance to antibiotics in bacteria: A systems biology perspective. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 253–267. [Google Scholar] [CrossRef]
- Egorova, D.A.; Solovyev, A.I.; Polyakov, N.B.; Danilova, K.V.; Scherbakova, A.A.; Kravtsov, I.N.; Dmitrieva, M.A.; Rykova, V.S.; Tutykhina, I.L.; Romanova, Y.M.; et al. Biofilm matrix proteome of clinical strain of P. aeruginosa isolated from bronchoalveolar lavage of patient in intensive care unit. Microb. Pathog. 2022, 170, 105714. [Google Scholar] [CrossRef]
- Barken, K.B.; Pamp, S.J.; Yang, L.; Gjermansen, M.; Bertrand, J.J.; Klausen, M.; Givskov, M.; Whitchurch, C.B.; Engel, J.N.; Tolker-Nielsen, T. Roles of type IV pili, flagellum-mediated motility and extracellular DNA in the formation of mature multicellular structures in Pseudomonas aeruginosa biofilms. Environ. Microbiol. 2008, 10, 2331–2343. [Google Scholar] [CrossRef]
- Mugunthan, S.; Wong, L.L.; Winnerdy, F.R.; Summers, S.; Bin Ismail, M.H.; Foo, Y.H.; Jaggi, T.K.; Meldrum, O.W.; Tiew, P.Y.; Chotirmall, S.H. RNA is a key component of extracellular DNA networks in Pseudomonas aeruginosa biofilms. Nat. Commun. 2023, 14, 7772. [Google Scholar] [CrossRef]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial biofilms. Annu. Rev. Microbial. J. 1995, 49, 71l–745. [Google Scholar] [CrossRef] [PubMed]
- Anderl, J.N.; Franklin, M.J.; Stewart, P.S. Role of Antibiotic Penetration Limitation in Klebsiella pneumoniae Biofilm Resistance to Ampicillin and Ciprofloxacin. Antimicrob. Agents Chemother. 2000, 44, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, S.Y.; Chan, S.Y.; Chua, S.L. Biofilm matrix cloaks bacterial quorum sensing chemoattractants from predator detection. ISME J. 2022, 16, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Liu, S.Y.; Seng, Z.; Chua, S.L. Biofilm matrix disrupts nematode motility and predatory behavior. ISME J. 2021, 15, 260–269. [Google Scholar] [CrossRef]
- Liu, Y.S.; Zhang, C.; Khoo, B.L.; Hao, P.; Chua, S.L. Dual-species proteomics and targeted intervention of animal-pathogen interactions. J. Adv. Res. 2024, in press. [CrossRef]
- Sarkar, S. Release mechanisms and molecular interactions of Pseudomonas aeruginosa extracellular DNA. Appl. Microbiol. Biotechnol. 2020, 104, 6549–6564. [Google Scholar] [CrossRef]
- Zhang, X.; Li, J.; Yao, M.C.; Fan, W.Y.; Yang, C.W.; Yuan, L.; Sheng, G.P. Unrecognized Contributions of Dissolved Organic Matter Inducing Photodamages to the Decay of Extracellular DNA in Waters. Environ. Sci. Technol. 2020, 54, 1614–1622. [Google Scholar] [CrossRef]
- Flemming, H.C.; van Hullebusch, E.D.; Neu, T.R.; Nielsen, P.H.; Seviour, T.; Stoodley, P.; Wingender, J.; Wuertz, S. The biofilm matrix: Multitasking in a shared space. Nat. Rev. Microbiol. 2023, 21, 70–86. [Google Scholar] [CrossRef]
- Okshevsky, M.; Meyer, R.L. The role of extracellular DNA in the establishment, maintenance and perpetuation of bacterial biofilms. Crit. Rev. Microbiol. 2015, 41, 341–352. [Google Scholar] [CrossRef]
- Fuxman Bass, J.I.; Russo, D.M.; Gabelloni, M.L.; Geffner, J.R.; Giordano, M.; Catalano, M.; Zorreguieta, A.; Trevani, A.S. Extracellular DNA: A major proinflammatory component of Pseudomonas aeruginosa biofilms. J. Immunol. 2010, 184, 6386–6395. [Google Scholar] [CrossRef]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.K.; Brannon, M.K.; Stevens, L.; Johansen, H.K.; Selgrade, S.E.; Miller, S.I.; Høiby, N.; Moskowitz, S.M. PhoQ mutations promote lipid A modification and polymyxin resistance of Pseudomonas aeruginosa found in colistin-treated cystic fibrosis patients. Antimicrob. Agents Chemother. 2011, 55, 5761–5769. [Google Scholar] [CrossRef] [PubMed]
- Chua, S.L.; Tan, S.Y.; Rybtke, M.T.; Chen, Y.; Rice, S.A.; Kjelleberg, S.; Tolker-Nielsen, T.; Yang, L.; Givskov, M. Bis-(3′-5′)-cyclic dimeric GMP regulates antimicrobial peptide resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.K.; Storek, K.M.; Ledvina, H.E.; Coulon, C.; Marmont, L.S.; Sadovskaya, I.; Secor, P.R.; Tseng, B.S.; Scian, M.; Filloux, A.; et al. Pel is a cationic exopolysaccharide that cross-links extracellular DNA in the Pseudomonas aeruginosa biofilm matrix. Proc. Natl. Acad. Sci. USA 2015, 112, 11353–11358. [Google Scholar] [CrossRef]
- Wang, S.; Liu, X.; Liu, H.; Zhang, L.; Guo, Y.; Yu, S.; Wozniak, D.J.; Ma, L.Z. The exopolysaccharide Psl-eDNA interaction enables the formation of a biofilm skeleton in Pseudomonas aeruginosa. Environ. Microbiol. Rep. 2015, 7, 330–340. [Google Scholar] [CrossRef]
- Rasamiravaka, T.; Labtani, Q.; Duez, P.; El Jaziri, M. The formation of biofilms by Pseudomonas aeruginosa: A review of the natural and synthetic compounds interfering with control mechanisms. BioMed Res. Int. 2015, 2015, 759348. [Google Scholar] [CrossRef]
- Rasamiravaka, T.; El Jaziri, M. Quorum-Sensing Mechanisms and Bacterial Response to Antibiotics in P. aeruginosa. Curr. Microbiol. 2016, 73, 747–753. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Givskov, M. The role of quorum sensing in the pathogenicity of the cunning aggressor Pseudomonas aeruginosa. Anal. Bioanal. Chem. 2007, 387, 409–414. [Google Scholar] [CrossRef]
- Rasamiravaka, T.; Vandeputte, O.M.; Pottier, L.; Huet, J.; Rabemanantsoa, C.; Kiendrebeogo, M.; Andriantsimahavandy, A.; Rasamindrakotroka, A.; Stévigny, C.; Duez, P.; et al. Pseudomonas aeruginosa Biofilm Formation and Persistence, along with the Production of Quorum Sensing-Dependent Virulence Factors, Are Disrupted by a Triterpenoid Coumarate Ester Isolated from Dalbergia trichocarpa, a Tropical Legume. PLoS ONE 2015, 10, e0132791. [Google Scholar] [CrossRef]
- Wilder, C.N.; Diggle, S.P.; Schuster, M. Cooperation and cheating in Pseudomonas aeruginosa: The roles of the las, rhl and pqs quorum-sensing systems. ISME J. 2011, 5, 1332–1343. [Google Scholar] [CrossRef]
- Valentini, M.; Filloux, A. Multiple roles of c-di-GMP signaling in bacterial pathogenesis. Annu. Rev. Microbiol. 2019, 73, 387–406. [Google Scholar] [CrossRef] [PubMed]
- Ha, D.-G.; O’Toole, G.A. c-di-GMP and its effects on biofilm formation and dispersion: A Pseudomonas aeruginosa review. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Chua, S.L.; Liu, Y.; Yam, J.K.; Chen, Y.; Vejborg, R.M.; Tan, B.G.; Kjelleberg, S.; Tolker-Nielsen, T.; Givskov, M.; Yang, L. Dispersed cells represent a distinct stage in the transition from bacterial biofilm to planktonic lifestyles. Nat. Commun. 2014, 5, 4462. [Google Scholar] [CrossRef] [PubMed]
- Lin Chua, S.; Liu, Y.; Li, Y.; Jun Ting, H.; Kohli, G.S.; Cai, Z.; Suwanchaikasem, P.; Kau Kit Goh, K.; Pin Ng, S.; Tolker-Nielsen, T.; et al. Reduced Intracellular c-di-GMP Content Increases Expression of Quorum Sensing-Regulated Genes in Pseudomonas aeruginosa. Front. Cell. Infect. Microbiol. 2017, 7, 451. [Google Scholar] [CrossRef]
- Chua, S.L.; Ding, Y.; Liu, Y.; Cai, Z.; Zhou, J.; Swarup, S.; Drautz-Moses, D.I.; Schuster, S.C.; Kjelleberg, S.; Givskov, M.; et al. Reactive oxygen species drive evolution of pro-biofilm variants in pathogens by modulating cyclic-di-GMP levels. Open Biol. 2016, 6, 160162. [Google Scholar] [CrossRef]
- Chua, S.L.; Hultqvist, L.D.; Yuan, M.; Rybtke, M.; Nielsen, T.E.; Givskov, M.; Tolker-Nielsen, T.; Yang, L. In vitro and in vivo generation and characterization of Pseudomonas aeruginosa biofilm–dispersed cells via c-di-GMP manipulation. Nat. Protoc. 2015, 10, 1165–1180. [Google Scholar] [CrossRef]
- Michiels, J.E.; Van den Bergh, B.; Verstraeten, N.; Michiels, J. Molecular mechanisms and clinical implications of bacterial persistence. Drug Resist. Updates 2016, 29, 76–89. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef]
- Van den Bergh, B.; Fauvart, M.; Michiels, J. Formation, physiology, ecology, evolution and clinical importance of bacterial persisters. FEMS Microbiol. Rev. 2017, 41, 219–251. [Google Scholar] [CrossRef]
- Wang, X.; Wood, T.K. Toxin-Antitoxin Systems Influence Biofilm and Persister Cell Formation and the General Stress Response. Appl. Environ. Microbiol. 2011, 77, 5577–5583. [Google Scholar] [CrossRef]
- Van Melderen, L.; Saavedra De Bast, M. Bacterial toxin–antitoxin systems: More than selfish entities? PLoS Genet. 2009, 5, e1000437. [Google Scholar] [CrossRef] [PubMed]
- Hazan, R.; Engelberg-Kulka, H. Escherichia coli mazEF-mediated cell death as a defense mechanism that inhibits the spread of phage P1. Mol. Genet. Genom. 2004, 272, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Horcajada Juan, P.; Montero, M.; Oliver, A.; Sorlí, L.; Luque, S.; Gómez-Zorrilla, S.; Benito, N.; Grau, S. Epidemiology and Treatment of Multidrug-Resistant and Extensively Drug-Resistant Pseudomonas aeruginosa Infections. Clin. Microbiol. Rev. 2019, 32, e00031-19. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.W.S.; Ma, Y.; Deng, Y.; Khoo, B.L.; Chua, S.L. Bacterial Iron Siderophore Drives Tumor Survival and Ferroptosis Resistance in a Biofilm-Tumor Spheroid Coculture Model. Adv. Sci. 2024, 11, 2404467. [Google Scholar] [CrossRef]
- Wang, S.; Chan, S.Y.; Deng, Y.; Khoo, B.L.; Chua, S.L. Oxidative stress induced by Etoposide anti-cancer chemotherapy drives the emergence of tumor-associated bacteria resistance to fluoroquinolones. J. Adv. Res. 2024, 55, 33–44. [Google Scholar] [CrossRef]
- Deng, Y.; Liu, S.Y.; Chua, S.L.; Khoo, B.L. The effects of biofilms on tumor progression in a 3D cancer-biofilm microfluidic model. Biosens. Bioelectron. 2021, 180, 113113. [Google Scholar] [CrossRef]
- Deng, Y.; Fu, Y.; Chua, S.L.; Khoo, B.L. Biofilm Potentiates Cancer-Promoting Effects of Tumor-Associated Macrophages in a 3D Multi-Faceted Tumor Model. Small 2023, 19, 2205904. [Google Scholar] [CrossRef]
- Arca-Suárez, J.; Fraile-Ribot, P.; Vázquez-Ucha, J.C.; Cabot, G.; Martínez-Guitián, M.; Lence, E.; González-Bello, C.; Beceiro, A.; Rodríguez-Iglesias, M.; Galán-Sánchez, F. Challenging antimicrobial susceptibility and evolution of resistance (OXA-681) during treatment of a long-term nosocomial infection caused by a Pseudomonas aeruginosa ST175 clone. Antimicrob. Agents Chemother. 2019, 63, e01110-19. [Google Scholar] [CrossRef]
- Ma, Y.; Deng, Y.; Hua, H.; Khoo, B.L.; Chua, S.L. Distinct bacterial population dynamics and disease dissemination after biofilm dispersal and disassembly. ISME J. 2023, 17, 1290–1302. [Google Scholar] [CrossRef]
- Yu, M.; Chua, S.L. Demolishing the great wall of biofilms in Gram-negative bacteria: To disrupt or disperse? Med. Res. Rev. 2020, 40, 1103–1116. [Google Scholar] [CrossRef]
- Chi, Y.; Xu, J.; Bai, N.; Liang, B.; Cai, Y. The efficacy and safety of Ceftolozane-Tazobactam in the treatment of GNB infections: A systematic review and meta-analysis of clinical studies. Expert Rev. Anti-Infect. Ther. 2023, 21, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Guzek, A.; Rybicki, Z.; Tomaszewski, D. An Analysis of the Type and Antimicrobial Resistance of Carbapenemase-Producing Enterobacteriaceae Isolated at the Military Institute of Medicine in Warsaw, Poland, 2009–2016. Jundishapur J. Microbiol. 2019, 12, 1–6. [Google Scholar] [CrossRef]
- van Duin, D.; Bonomo, R.A. Ceftazidime/Avibactam and Ceftolozane/Tazobactam: Second-generation β-Lactam/β-Lactamase Inhibitor Combinations. Clin. Infect. Dis. 2016, 63, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Sato, T.; Ota, M.; Takemura, M.; Nishikawa, T.; Toba, S.; Kohira, N.; Miyagawa, S.; Ishibashi, N.; Matsumoto, S.; et al. In Vitro Antibacterial Properties of Cefiderocol, a Novel Siderophore Cephalosporin, against Gram-Negative Bacteria. Antimicrob. Agents Chemother. 2018, 62, 1228–1247. [Google Scholar] [CrossRef]
- Mikhail, S.; Singh, N.B.; Kebriaei, R.; Rice, S.A.; Stamper, K.C.; Castanheira, M.; Rybak, M.J. Evaluation of the Synergy of Ceftazidime-Avibactam in Combination with Meropenem, Amikacin, Aztreonam, Colistin, or Fosfomycin against Well-Characterized Multidrug-Resistant Klebsiella pneumoniae and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e00779-19. [Google Scholar] [CrossRef]
- Torres, D.A.; Seth-Smith, H.M.B.; Joosse, N.; Lang, C.; Dubuis, O.; Nüesch-Inderbinen, M.; Hinic, V.; Egli, A. Colistin resistance in Gram-negative bacteria analysed by five phenotypic assays and inference of the underlying genomic mechanisms. BMC Microbiol. 2021, 21, 321. [Google Scholar] [CrossRef]
- Montini, G.; Tessitore, A.; Console, K.; Ronfani, L.; Barbi, E.; Pennesi, M. Short Oral Antibiotic Therapy for Pediatric Febrile Urinary Tract Infections: A Randomized Trial. Pediatrics 2024, 153, e2023062598. [Google Scholar] [CrossRef]
- Noronha, A.A.; Domingues, G.R.; de Souza, G.C.; Nau, A.L.; Lo, D.S. Short- versus standard-course antibiotic therapy for urinary tract infection in children: A systematic review and meta-analysis. Pediatr. Nephrol. 2024. [Google Scholar] [CrossRef]
- Lashkar, M.O.; Nahata, M.C. Antimicrobial Pharmacotherapy Management of Urinary Tract Infections in Pediatric Patients. J. Pharm. Technol. 2018, 34, 62–81. [Google Scholar] [CrossRef]
- Autore, G.; Bernardi, L.; La Scola, C.; Ghidini, F.; Marchetti, F.; Pasini, A.; Pierantoni, L.; Castellini, C.; Gatti, C.; Malaventura, C.; et al. Management of Pediatric Urinary Tract Infections: A Delphi Study. Antibiotics 2022, 11, 1122. [Google Scholar] [CrossRef]
- Debarbieux, L.; Leduc, D.; Maura, D.; Morello, E.; Criscuolo, A.; Grossi, O.; Balloy, V.; Touqui, L. Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J. Infect. Dis. 2010, 201, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Morello, E.; Saussereau, E.; Maura, D.; Huerre, M.; Touqui, L.; Debarbieux, L. Pulmonary bacteriophage therapy on Pseudomonas aeruginosa cystic fibrosis strains: First steps towards treatment and prevention. PLoS ONE 2011, 6, e16963. [Google Scholar] [CrossRef] [PubMed]
- Khawaldeh, A.; Morales, S.; Dillon, B.; Alavidze, Z.; Ginn, A.; Thomas, L.; Chapman, S.; Dublanchet, A.; Smithyman, A.; Iredell, J. Bacteriophage therapy for refractory Pseudomonas aeruginosa urinary tract infection. J. Med. Microbiol. 2011, 60, 1697–1700. [Google Scholar] [CrossRef] [PubMed]
- Marza, J.S.; Soothill, J.; Boydell, P.; Collyns, T. Multiplication of therapeutically administered bacteriophages in Pseudomonas aeruginosa infected patients. Burns 2006, 32, 644–646. [Google Scholar] [CrossRef]
- Aslam, S.; Courtwright, A.M.; Koval, C.; Lehman, S.M.; Morales, S.; Furr, C.-L.L.; Rosas, F.; Brownstein, M.J.; Fackler, J.R.; Sisson, B.M. Early clinical experience of bacteriophage therapy in 3 lung transplant recipients. Am. J. Transplant. 2019, 19, 2631–2639. [Google Scholar] [CrossRef]
- Marashi, S.M.A.; Nikkhahi, F.; Hamedi, D.; Shahbazi, G. Isolation, Characterization and in vitro Evaluation of Specific Bacteriophages Targeting Extensive Drug Resistance Strains of Pseudomonas aeruginosa Isolated from Septic Burn Wounds. Infect. Chemother. 2022, 54, 153–164. [Google Scholar] [CrossRef]
- Luong, T.; Salabarria, A.-C.; Roach, D.R. Phage therapy in the resistance era: Where do we stand and where are we going? Clin. Ther. 2020, 42, 1659–1680. [Google Scholar] [CrossRef]
- Zalewska-Piątek, B. Phage Therapy—Challenges, Opportunities and Future Prospects. Pharmaceuticals 2023, 16, 1638. [Google Scholar] [CrossRef]
- Kunisch, F.; Campobasso, C.; Wagemans, J.; Yildirim, S.; Chan, B.K.; Schaudinn, C.; Lavigne, R.; Turner, P.E.; Raschke, M.J.; Trampuz, A.; et al. Targeting Pseudomonas aeruginosa biofilm with an evolutionary trained bacteriophage cocktail exploiting phage resistance trade-offs. Nat. Commun. 2024, 15, 8572. [Google Scholar] [CrossRef]
- Chegini, Z.; Khoshbayan, A.; Taati Moghadam, M.; Farahani, I.; Jazireian, P.; Shariati, A. Bacteriophage therapy against Pseudomonas aeruginosa biofilms: A review. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 45. [Google Scholar] [CrossRef]
- Mion, S.; Rémy, B.; Plener, L.; Brégeon, F.; Chabrière, E.; Daudé, D. Quorum Quenching Lactonase Strengthens Bacteriophage and Antibiotic Arsenal Against Pseudomonas aeruginosa Clinical Isolates. Front. Microbiol. 2019, 10, 2049. [Google Scholar] [CrossRef] [PubMed]
- Leitner, L.; Ujmajuridze, A.; Chanishvili, N.; Goderdzishvili, M.; Chkonia, I.; Rigvava, S.; Chkhotua, A.; Changashvili, G.; McCallin, S.; Schneider, M.P. Intravesical bacteriophages for treating urinary tract infections in patients undergoing transurethral resection of the prostate: A randomised, placebo-controlled, double-blind clinical trial. Lancet Infect. Dis. 2021, 21, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Vaitekenas, A.; Tai, A.S.; Ramsay, J.P.; Stick, S.M.; Kicic, A. Pseudomonas aeruginosa Resistance to Bacteriophages and Its Prevention by Strategic Therapeutic Cocktail Formulation. Antibiotics 2021, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, S.O.; Sahu, S.K.; Yadav, V.K.; Rathod, M.C.; Patel, D.; Sahoo, D.K.; Patel, A. Bacteriophages as a potential substitute for antibiotics: A comprehensive review. Cell Biochem. Funct. 2024, 42, e4022. [Google Scholar] [CrossRef]
- Paul, V.D.; Sundarrajan, S.; Rajagopalan, S.S.; Hariharan, S.; Kempashanaiah, N.; Padmanabhan, S.; Sriram, B.; Ramachandran, J. Lysis-deficient phages as novel therapeutic agents for controlling bacterial infection. BMC Microbiol. 2011, 11, 195. [Google Scholar] [CrossRef]
- Waters, E.M.; Neill, D.R.; Kaman, B.; Sahota, J.S.; Clokie, M.R.J.; Winstanley, C.; Kadioglu, A. Phage therapy is highly effective against chronic lung infections with Pseudomonas aeruginosa. Thorax 2017, 72, 666–667. [Google Scholar] [CrossRef]
- Fu, W.; Forster, T.; Mayer, O.; Curtin, J.J.; Lehman, S.M.; Donlan, R.M. Bacteriophage cocktail for the prevention of biofilm formation by Pseudomonas aeruginosa on catheters in an in vitro model system. Antimicrob. Agents Chemother. 2010, 54, 397–404. [Google Scholar] [CrossRef]
- Hagens, S.; Habel, A.; von Ahsen, U.; von Gabain, A.; Bläsi, U. Therapy of experimental pseudomonas infections with a nonreplicating genetically modified phage. Antimicrob. Agents Chemother. 2004, 48, 3817–3822. [Google Scholar] [CrossRef]
- Henriksen, K.; Rørbo, N.; Rybtke, M.L.; Martinet, M.G.; Tolker-Nielsen, T.; Høiby, N.; Middelboe, M.; Ciofu, O. P. aeruginosa flow-cell biofilms are enhanced by repeated phage treatments but can be eradicated by phage–ciprofloxacin combination: —Monitoring the phage–P. aeruginosa biofilms interactions. Pathog. Dis. 2019, 77, ftz011. [Google Scholar] [CrossRef]
- Nang, S.C.; Lin, Y.W.; Petrovic Fabijan, A.; Chang, R.Y.K.; Rao, G.G.; Iredell, J.; Chan, H.K.; Li, J. Pharmacokinetics/pharmacodynamics of phage therapy: A major hurdle to clinical translation. Clin. Microbiol. Infect. 2023, 29, 702–709. [Google Scholar] [CrossRef]
- Dąbrowska, K.; Abedon, S.T. Pharmacologically aware phage therapy: Pharmacodynamic and pharmacokinetic obstacles to phage antibacterial action in animal and human bodies. Microbiol. Mol. Biol. Rev. 2019, 83, e00012-19. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowska, K. Phage therapy: What factors shape phage pharmacokinetics and bioavailability? Systematic and critical review. Med. Res. Rev. 2019, 39, 2000–2025. [Google Scholar] [CrossRef] [PubMed]
- Torres-Barceló, C. Phage Therapy Faces Evolutionary Challenges. Viruses 2018, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- Krut, O.; Bekeredjian-Ding, I. Contribution of the Immune Response to Phage Therapy. J. Immunol. 2018, 200, 3037–3044. [Google Scholar] [CrossRef]
- Abedon, S.T.; Thomas-Abedon, C. Phage therapy pharmacology. Curr. Pharm. Biotechnol. 2010, 11, 28–47. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Wallin, M.; Lin, Y.; Leung, S.S.Y.; Wang, H.; Morales, S.; Chan, H.-K. Phage therapy for respiratory infections. Adv. Drug Deliv. Rev. 2018, 133, 76–86. [Google Scholar] [CrossRef]
- Jończyk, E.; Kłak, M.; Międzybrodzki, R.; Górski, A. The influence of external factors on bacteriophages—Review. Folia Microbiol. 2011, 56, 191–200. [Google Scholar] [CrossRef]
- Pagava, K.; Gachechiladze, K.; Korinteli, I.; Dzuliashvili, M.; Alavidze, Z.; Hoyle, N.; Metskhvarishvili, G. What happens when the child gets bacteriophage per os? Georgian Med. News 2011, 196–197, 101–105. [Google Scholar]
- Koskella, B.; Brockhurst, M.A. Bacteria–phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef]
- Harper, D.; Enright, M. Bacteriophages for the treatment of Pseudomonas aeruginosa infections. J. Appl. Microbiol. 2011, 111, 1–7. [Google Scholar] [CrossRef]
- Abedon, S.T.; Danis-Wlodarczyk, K.M.; Wozniak, D.J. Phage cocktail development for bacteriophage therapy: Toward improving spectrum of activity breadth and depth. Pharmaceuticals 2021, 14, 1019. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, F.; Piccardi, P.; Mancini, S.; Gabard, J.; Moreillon, P.; Entenza, J.M.; Resch, G.; Que, Y.-A. Synergistic interaction between phage therapy and antibiotics clears Pseudomonas aeruginosa infection in endocarditis and reduces virulence. J. Infect. Dis. 2017, 215, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Sistrom, M.; Wertz, J.E.; Kortright, K.E.; Narayan, D.; Turner, P.E. Phage selection restores antibiotic sensitivity in MDR Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 26717. [Google Scholar] [CrossRef] [PubMed]
- Bavaro, D.F.; Romanelli, F.; Stolfa, S.; Belati, A.; Diella, L.; Ronga, L.; Fico, C.; Monno, L.; Mosca, A.; Saracino, A. Recurrent neurosurgical site infection by extensively drug-resistant P. aeruginosa treated with cefiderocol: A case report and literature review. Infect. Dis. 2021, 53, 206–211. [Google Scholar] [CrossRef]
- Cesta, N.; Pini, M.; Mulas, T.; Materazzi, A.; Ippolito, E.; Wagemans, J.; Kutateladze, M.; Fontana, C.; Sarmati, L.; Tavanti, A. Application of phage therapy in a case of a chronic hip-prosthetic joint infection due to Pseudomonas aeruginosa: An Italian real-life experience and in vitro analysis. Open Forum Infect. Dis. 2023, 10, ofad051. [Google Scholar] [CrossRef]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Lazzaro, B.P.; Zasloff, M.; Rolff, J. Antimicrobial peptides: Application informed by evolution. Science 2020, 368, eaau5480. [Google Scholar] [CrossRef]
- Zhang, K.; Li, X.; Yu, C.; Wang, Y. Promising Therapeutic Strategies Against Microbial Biofilm Challenges. Front. Cell. Infect. Microbiol. 2020, 10, 359. [Google Scholar] [CrossRef]
- Juárez-López, D.; Morales-Ruiz, E.; Herrera-Zúñiga, L.D.; González-Carrera, Z.; Cuevas-Reyes, E.; Corzo, G.; Schcolnik-Cabrera, A.; Villegas, E. The Resilience of Pseudomonas aeruginosa to Antibiotics and the Designing of Antimicrobial Peptides to Overcome Microbial Resistance. Curr. Med. Chem. 2022, 30, 72–103. [Google Scholar] [CrossRef]
- Roversi, D.; Troiano, C.; Salnikov, E.; Giordano, L.; Riccitelli, F.; De Zotti, M.; Casciaro, B.; Loffredo, M.R.; Park, Y.; Formaggio, F. Effects of antimicrobial peptides on membrane dynamics: A comparison of fluorescence and NMR experiments. Biophys. Chem. 2023, 300, 107060. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Sugishita, K.-i.; Ishibe, N.; Ueha, M.; Nakata, S.; Miyajima, K.; Epand, R.M. Relationship of Membrane Curvature to the Formation of Pores by Magainin 2. Biochemistry 1998, 37, 11856–11863. [Google Scholar] [CrossRef] [PubMed]
- Aminnia, P.; Niknafs, A.S.; Doustdar, F. From Bench to Bedside: A Comprehensive Study on Pardaxin Peptide’s Antimicrobial Effect on Escherichia coli, Including Clinical Isolates. Arch. Razi Inst. 2024, 79, 963–966. [Google Scholar]
- Shai, Y. Pardaxin: Channel formation by a shark repellant peptide from fish. Toxicology 1994, 87, 109–129. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Yoneyama, F.; Imura, Y.; Ohno, K.; Zendo, T.; Nakayama, J.; Matsuzaki, K.; Sonomoto, K. Peptide-lipid huge toroidal pore, a new antimicrobial mechanism mediated by a lactococcal bacteriocin, lacticin Q. Antimicrob. Agents Chemother. 2009, 53, 3211–3217. [Google Scholar] [CrossRef]
- Anbarasu, A. Antimicrobial peptides as Immunomodulators and Antimycobacterial agents to combat Mycobacterium tuberculosis: A critical review. Probiotics Antimicrob. Proteins 2023, 15, 1539–1566. [Google Scholar]
- Melo, M.N.; Ferre, R.; Castanho, M.A.R.B. Antimicrobial peptides: Linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 2009, 7, 245–250. [Google Scholar] [CrossRef]
- Wu, M.; Maier, E.; Benz, R.; Hancock, R.E.W. Mechanism of Interaction of Different Classes of Cationic Antimicrobial Peptides with Planar Bilayers and with the Cytoplasmic Membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar] [CrossRef]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.; Rehm, B.H.; Hancock, R.E. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef]
- Dean, S.N.; Bishop, B.M.; Van Hoek, M.L. Susceptibility of Pseudomonas aeruginosa biofilm to alpha-helical peptides: D-enantiomer of LL-37. Front. Microbiol. 2011, 2, 128. [Google Scholar] [CrossRef]
- De La Fuente-Núñez, C.; Korolik, V.; Bains, M.; Nguyen, U.; Breidenstein, E.B.; Horsman, S.; Lewenza, S.; Burrows, L.; Hancock, R.E. Inhibition of bacterial biofilm formation and swarming motility by a small synthetic cationic peptide. Antimicrob. Agents Chemother. 2012, 56, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Wolz, C.; Geiger, T.; Goerke, C. The synthesis and function of the alarmone (p) ppGpp in firmicutes. Int. J. Med. Microbiol. 2010, 300, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Gorr, S.U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-Q.; Kyung, S.M.; Kim, S.; Kim, G.; Lee, S.Y.; Yoo, H.S. Effects of synthetic peptide RP557 and its origin, LL-37, on carbapenem-resistant Pseudomonas aeruginosa. Microbiol. Spectr. 2023, 11, e00430-23. [Google Scholar] [CrossRef]
- Luca, V.; Stringaro, A.; Colone, M.; Pini, A.; Mangoni, M.L. Esculentin(1-21), an amphibian skin membrane-active peptide with potent activity on both planktonic and biofilm cells of the bacterial pathogen Pseudomonas aeruginosa. Cell. Mol. Life Sci. 2013, 70, 2773–2786. [Google Scholar] [CrossRef]
- Pletzer, D.; Hancock, R.E. Antibiofilm peptides: Potential as broad-spectrum agents. J. Bacteriol. 2016, 198, 2572–2578. [Google Scholar] [CrossRef]
- Memariani, M.; Memariani, H. Anti-biofilm effects of melittin: Lessons learned and the path ahead. Int. J. Pept. Res. Ther. 2024, 30, 29. [Google Scholar] [CrossRef]
- Gomes, D.; Santos, R.; Soares, R.S.; Reis, S.; Carvalho, S.; Rego, P.; Peleteiro, M.C.; Tavares, L.; Oliveira, M. Pexiganan in combination with nisin to control polymicrobial diabetic foot infections. Antibiotics 2020, 9, 128. [Google Scholar] [CrossRef]
- Yin, C.; Alam, M.Z.; Fallon, J.T.; Huang, W. Advances in development of novel therapeutic strategies against multi-drug resistant Pseudomonas aeruginosa. Antibiotics 2024, 13, 119. [Google Scholar] [CrossRef]
- Ramamourthy, G.; Vogel, H.J. Antibiofilm activity of lactoferrin-derived synthetic peptides against Pseudomonas aeruginosa PAO1. Biochem. Cell Biol. 2021, 99, 138–148. [Google Scholar] [CrossRef]
- Sahoo, A.; Swain, S.S.; Behera, A.; Sahoo, G.; Mahapatra, P.K.; Panda, S.K. Antimicrobial peptides derived from insects offer a novel therapeutic option to combat biofilm: A review. Front. Microbiol. 2021, 12, 661195. [Google Scholar] [CrossRef] [PubMed]
- Ali, W.; Elsahn, A.; Ting, D.S.; Dua, H.S.; Mohammed, I. Host defence peptides: A potent alternative to combat antimicrobial resistance in the era of the COVID-19 pandemic. Antibiotics 2022, 11, 475. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, Q.; Xu, T.; Malakar, P.K.; Zhu, Y.; Liu, J.; Zhao, Y.; Zhang, Z. Thanatin: A Promising Antimicrobial Peptide Targeting the Achilles’ Heel of Multidrug-Resistant Bacteria. Int. J. Mol. Sci. 2024, 25, 9496. [Google Scholar] [CrossRef] [PubMed]
- Ruffin, M.; Brochiero, E. Repair process impairment by Pseudomonas aeruginosa in epithelial tissues: Major features and potential therapeutic avenues. Front. Cell. Infect. Microbiol. 2019, 9, 182. [Google Scholar] [CrossRef]
- Greco, I.; Emborg, A.P.; Jana, B.; Molchanova, N.; Oddo, A.; Damborg, P.; Guardabassi, L.; Hansen, P.R. Characterization, mechanism of action and optimization of activity of a novel peptide-peptoid hybrid against bacterial pathogens involved in canine skin infections. Sci. Rep. 2019, 9, 3679. [Google Scholar] [CrossRef]
- Aoki, W.; Ueda, M. Characterization of Antimicrobial Peptides toward the Development of Novel Antibiotics. Pharmaceuticals 2013, 6, 1055. [Google Scholar] [CrossRef]
- Costa, F.; Teixeira, C.; Gomes, P.; Martins, M.C.L. Clinical Application of AMPs. In Antimicrobial Peptides: Basics for Clinical Application; Matsuzaki, K., Ed.; Springer: Singapore, 2019; pp. 281–298. [Google Scholar]
- Luo, Y.; Song, Y. Mechanism of Antimicrobial Peptides: Antimicrobial, Anti-Inflammatory and Antibiofilm Activities. Int. J. Mol. Sci. 2021, 22, 11401. [Google Scholar] [CrossRef]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Mwangi, J.; Kamau, P.M.; Thuku, R.C.; Lai, R. Design methods for antimicrobial peptides with improved performance. Zool. Res. 2023, 44, 1095–1114. [Google Scholar] [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- O’Loughlin, C.T.; Miller, L.C.; Siryaporn, A.; Drescher, K.; Semmelhack, M.F.; Bassler, B.L. A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc. Natl. Acad. Sci. USA 2013, 110, 17981–17986. [Google Scholar] [CrossRef] [PubMed]
- Castang, S.; Chantegrel, B.; Deshayes, C.; Dolmazon, R.; Gouet, P.; Haser, R.; Reverchon, S.; Nasser, W.; Hugouvieux-Cotte-Pattat, N.; Doutheau, A. N-Sulfonyl homoserine lactones as antagonists of bacterial quorum sensing. Bioorganic Med. Chem. Lett. 2004, 14, 5145–5149. [Google Scholar] [CrossRef] [PubMed]
- Kalia, V.C. Quorum sensing inhibitors: An overview. Biotechnol. Adv. 2013, 31, 224–245. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Y.-Y.; Chua, S.-L.; Chen, Y.; Rice, S.A.; Kjelleberg, S.; Nielsen, T.E.; Yang, L.; Givskov, M. Identification of Five Structurally Unrelated Quorum-Sensing Inhibitors of Pseudomonas aeruginosa from a Natural-Derivative Database. Antimicrob. Agents Chemother. 2013, 57, 5629–5641. [Google Scholar] [CrossRef]
- Jakobsen, T.H.; van Gennip, M.; Phipps, R.K.; Shanmugham, M.S.; Christensen, L.D.; Alhede, M.; Skindersoe, M.E.; Rasmussen, T.B.; Friedrich, K.; Uthe, F. Ajoene, a sulfur-rich molecule from garlic, inhibits genes controlled by quorum sensing. Antimicrob. Agents Chemother. 2012, 56, 2314–2325. [Google Scholar] [CrossRef]
- Fong, J.; Yuan, M.; Jakobsen, T.H.; Mortensen, K.T.; Delos Santos, M.M.S.; Chua, S.L.; Yang, L.; Tan, C.H.; Nielsen, T.E.; Givskov, M. Disulfide Bond-Containing Ajoene Analogues As Novel Quorum Sensing Inhibitors of Pseudomonas aeruginosa. J. Med. Chem. 2017, 60, 215–227. [Google Scholar] [CrossRef]
- Tan, S.Y.; Liu, Y.; Chua, S.L.; Vejborg, R.M.; Jakobsen, T.H.; Chew, S.C.; Li, Y.; Nielsen, T.E.; Tolker-Nielsen, T.; Yang, L.; et al. Comparative systems biology analysis to study the mode of action of the isothiocyanate compound Iberin on Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 6648–6659. [Google Scholar] [CrossRef]
- Bendary, M.M.; Ali, M.A.; Abdel Halim, A.S.; Boufahja, F.; Chaudhary, A.A.; Elkelish, A.; Soliman, R.H.; Hegazy, W.A. Investigating Sulforaphane’s anti-virulence and anti-quorum sensing properties against Pseudomonas aeruginosa. Front. Pharmacol. 2024, 15, 1406653. [Google Scholar] [CrossRef]
- Kumar, N.V.; Murthy, P.S.; Manjunatha, J.R.; Bettadaiah, B. Synthesis and quorum sensing inhibitory activity of key phenolic compounds of ginger and their derivatives. Food Chem. 2014, 159, 451–457. [Google Scholar] [CrossRef]
- Chakraborty, P.; Dastidar, D.G.; Paul, P.; Dutta, S.; Basu, D.; Sharma, S.R.; Basu, S.; Sarker, R.K.; Sen, A.; Sarkar, A. Inhibition of biofilm formation of Pseudomonas aeruginosa by caffeine: A potential approach for sustainable management of biofilm. Arch. Microbiol. 2020, 202, 623–635. [Google Scholar] [CrossRef]
- Vandeputte, O.M.; Kiendrebeogo, M.; Rajaonson, S.; Diallo, B.; Mol, A.; El Jaziri, M.; Baucher, M. Identification of catechin as one of the flavonoids from Combretum albiflorum bark extract that reduces the production of quorum-sensing-controlled virulence factors in Pseudomonas aeruginosa PAO1. Appl. Environ. Microbiol. 2010, 76, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, R.; Mondal, C.; Bera, R.; Chakraborty, S.; Barik, R.; Roy, P.; Kumar, A.; Yadav, K.K.; Choudhury, J.; Chaudhary, S.K. Antimicrobial properties of K alanchoe blossfeldiana: A focus on drug resistance with particular reference to quorum sensing-mediated bacterial biofilm formation. J. Pharm. Pharmacol. 2015, 67, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Husain, F.M.; Ahmad, I.; Asif, M.; Tahseen, Q. Influence of clove oil on certain quorum-sensing-regulated functions and biofilm of Pseudomonas aeruginosa and Aeromonas hydrophila. J. Biosci. 2013, 38, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Tang, W.S.; Liu, S.Y.; Khoo, B.L.; Chua, S.L. Juglone as a Natural Quorum Sensing Inhibitor against Pseudomonas aeruginosa pqs-Mediated Virulence and Biofilms. ACS Pharmacol. Transl. Sci. 2024, 7, 533–543. [Google Scholar] [CrossRef]
- Quecan, B.X.; Santos, J.T.; Rivera, M.L.; Hassimotto, N.M.; Almeida, F.A.; Pinto, U.M. Effect of quercetin rich onion extracts on bacterial quorum sensing. Front. Microbiol. 2019, 10, 867. [Google Scholar] [CrossRef]
- Gui, M.; Wu, R.; Liu, L.; Wang, S.; Zhang, L.; Li, P. Effects of quorum quenching by AHL lactonase on AHLs, protease, motility and proteome patterns in Aeromonas veronii LP-11. Int. J. Food Microbiol. 2017, 252, 61–68. [Google Scholar] [CrossRef]
- Camps, J.; Pujol, I.; Ballester, F.; Joven, J.; Simó, J.M. Paraoxonases as potential antibiofilm agents: Their relationship with quorum-sensing signals in Gram-negative bacteria. Antimicrob. Agents Chemother. 2011, 55, 1325–1331. [Google Scholar] [CrossRef]
- Mok, N.; Chan, S.Y.; Liu, S.Y.; Chua, S.L. Vanillin inhibits PqsR-mediated virulence in Pseudomonas aeruginosa. Food Funct. 2020, 11, 6496–6508. [Google Scholar] [CrossRef]
- Marin, S.D.L.; Xu, Y.; Meijler, M.M.; Janda, K.D. Antibody catalyzed hydrolysis of a quorum sensing signal found in Gram-negative bacteria. Bioorganic Med. Chem. Lett. 2007, 17, 1549–1552. [Google Scholar] [CrossRef]
- Nalca, Y.; Jänsch, L.; Bredenbruch, F.; Geffers, R.; Buer, J.; Häussler, S. Quorum-sensing antagonistic activities of azithromycin in Pseudomonas aeruginosa PAO1: A global approach. Antimicrob. Agents Chemother. 2006, 50, 1680–1688. [Google Scholar] [CrossRef]
- Chua, S.L.; Yam, J.K.H.; Hao, P.; Adav, S.S.; Salido, M.M.; Liu, Y.; Givskov, M.; Sze, S.K.; Tolker-Nielsen, T.; Yang, L. Selective labelling and eradication of antibiotic-tolerant bacterial populations in Pseudomonas aeruginosa biofilms. Nat. Commun. 2016, 7, 10750. [Google Scholar] [CrossRef] [PubMed]
- Shakour, N.; Taheri, E.; Rajabian, F.; Tarighi, S.; Soheili, V.; Hadizadeh, F. Evaluating the antivirulence effects of new thiazolidinedione compounds against Pseudomonas aeruginosa PAO1. Microb. Drug Resist. 2022, 28, 1003–1018. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Song, Z.; Hentzer, M.; Andersen, J.B.; Molin, S.; Givskov, M.; Høiby, N. Synthetic furanones inhibit quorum-sensing and enhance bacterial clearance in Pseudomonas aeruginosa lung infection in mice. J. Antimicrob. Chemother. 2004, 53, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Kajiyama, K.; Kita, T.; Takiguchi, N.; Kuroda, A.; Kato, J.; Ohtake, H. The synthesis of optically pure enantiomers of N-acyl-homoserine lactone autoinducers and their analogues. Chem. Lett. 2001, 30, 314–315. [Google Scholar] [CrossRef]
- Deb Adhikari, M.; Roychowdhury, A.; Tiwary, B.K. Quorum Quenching Enzymes: A Potent Alternative to Conventional Antibiotics. In Alternatives to Antibiotics: Recent Trends and Future Prospects; Springer: Berlin/Heidelberg, Germany, 2022; pp. 57–81. [Google Scholar]
- Haque, S.; Ahmad, F.; Dar, S.A.; Jawed, A.; Mandal, R.K.; Wahid, M.; Lohani, M.; Khan, S.; Singh, V.; Akhter, N. Developments in strategies for Quorum Sensing virulence factor inhibition to combat bacterial drug resistance. Microb. Pathog. 2018, 121, 293–302. [Google Scholar] [CrossRef]
- García-Contreras, R.; Nunez-Lopez, L.; Jasso-Chávez, R.; Kwan, B.W.; Belmont, J.A.; Rangel-Vega, A.; Maeda, T.; Wood, T.K. Quorum sensing enhancement of the stress response promotes resistance to quorum quenching and prevents social cheating. ISME J. 2015, 9, 115–125. [Google Scholar] [CrossRef]
- García-Contreras, R.; Martínez-Vázquez, M.; Velázquez Guadarrama, N.; Villegas Pañeda, A.G.; Hashimoto, T.; Maeda, T.; Quezada, H.; Wood, T.K. Resistance to the quorum-quenching compounds brominated furanone C-30 and 5-fluorouracil in Pseudomonas aeruginosa clinical isolates. Pathog. Dis. 2013, 68, 8–11. [Google Scholar] [CrossRef]
- Maeda, T.; García-Contreras, R.; Pu, M.; Sheng, L.; Garcia, L.R.; Tomás, M.; Wood, T.K. Quorum quenching quandary: Resistance to antivirulence compounds. ISME J. 2012, 6, 493–501. [Google Scholar] [CrossRef]
- Tomás, M.; Doumith, M.; Warner, M.; Turton, J.F.; Beceiro, A.; Bou, G.; Livermore, D.M.; Woodford, N. Efflux pumps, OprD porin, AmpC β-lactamase, and multiresistance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Antimicrob. Agents Chemother. 2010, 54, 2219–2224. [Google Scholar] [CrossRef]
- Köhler, T.; Perron, G.G.; Buckling, A.; Van Delden, C. Quorum sensing inhibition selects for virulence and cooperation in Pseudomonas aeruginosa. PLoS Pathog. 2010, 6, e1000883. [Google Scholar] [CrossRef]
- Hoffmann, N.; Lee, B.; Hentzer, M.; Rasmussen, T.B.; Song, Z.; Johansen, H.K.; Givskov, M.; Høiby, N. Azithromycin blocks quorum sensing and alginate polymer formation and increases the sensitivity to serum and stationary-growth-phase killing of Pseudomonas aeruginosa and attenuates chronic P. aeruginosa lung infection in Cftr−/− mice. Antimicrob. Agents Chemother. 2007, 51, 3677–3687. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Liang, J.; Tang, Z.; Ma, R.; Peng, H.; Huang, Z. Role of the luxS gene in initial biofilm formation by Streptococcus mutans. J. Mol. Microbiol. Biotechnol. 2015, 25, 60–68. [Google Scholar] [PubMed]
- D’Argenio, D.A.; Wu, M.; Hoffman, L.R.; Kulasekara, H.D.; Déziel, E.; Smith, E.E.; Nguyen, H.; Ernst, R.K.; Larson Freeman, T.J.; Spencer, D.H. Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Mol. Microbiol. 2007, 64, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Yang, X.; Zeng, Q.; Zhang, Y.; Li, H.; Yan, C.; Li, J.S.; Liu, H.; Du, L.; Wu, Y. Evolution of lasR mutants in polymorphic Pseudomonas aeruginosa populations facilitates chronic infection of the lung. Nat. Commun. 2023, 14, 5976. [Google Scholar] [CrossRef]
- Mould, D.L. Causes and Consequences of Lasr Mutant Selection in Pseudomonas aeruginosa Populations. Ph.D. Thesis, Dartmouth College, Hanover, NH, USA, 2023. [Google Scholar]
- Azimi, S.; Roberts, A.E.; Peng, S.; Weitz, J.S.; McNally, A.; Brown, S.P.; Diggle, S.P. Allelic polymorphism shapes community function in evolving Pseudomonas aeruginosa populations. ISME J. 2020, 14, 1929–1942. [Google Scholar] [CrossRef]
- Kalia, V.C.; Wood, T.K.; Kumar, P. Evolution of resistance to quorum-sensing inhibitors. Microb. Ecol. 2014, 68, 13–23. [Google Scholar] [CrossRef]
- Kundar, R.; Gokarn, K. CRISPR-Cas system: A tool to eliminate drug-resistant gram-negative bacteria. Pharmaceuticals 2022, 15, 1498. [Google Scholar] [CrossRef]
- Nishita, M.; Park, S.-Y.; Nishio, T.; Kamizaki, K.; Wang, Z.; Tamada, K.; Takumi, T.; Hashimoto, R.; Otani, H.; Pazour, G.J. Ror2 signaling regulates Golgi structure and transport through IFT20 for tumor invasiveness. Sci. Rep. 2017, 7, 1. [Google Scholar] [CrossRef]
- Kiga, K.; Tan, X.-E.; Ibarra-Chávez, R.; Watanabe, S.; Aiba, Y.; Sato’o, Y.; Li, F.-Y.; Sasahara, T.; Cui, B.; Kawauchi, M. Development of CRISPR-Cas13a-based antimicrobials capable of sequence-specific killing of target bacteria. Nat. Commun. 2020, 11, 2934. [Google Scholar] [CrossRef]
- Selle, K.; Fletcher, J.R.; Tuson, H.; Schmitt, D.S.; McMillan, L.; Vridhambal, G.S.; Rivera, A.J.; Montgomery, S.A.; Fortier, L.-C.; Barrangou, R. In vivo targeting of Clostridioides difficile using phage-delivered CRISPR-Cas3 antimicrobials. mBio 2020, 11, e00019-20. [Google Scholar] [CrossRef]
- Kadkhoda, H.; Gholizadeh, P.; Samadi Kafil, H.; Ghotaslou, R.; Pirzadeh, T.; Ahangarzadeh Rezaee, M.; Nabizadeh, E.; Feizi, H.; Aghazadeh, M. Role of CRISPR-Cas systems and anti-CRISPR proteins in bacterial antibiotic resistance. Heliyon 2024, 10, e34692. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, Y.; Dong, N.; Shen, L.; Zhou, H.; Hu, Y.; Gu, D.; Chen, S.; Zhang, R.; Ji, Q. Application of CRISPR/Cas9-Based Genome Editing in Studying the Mechanism of Pandrug Resistance in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2019, 63, e00113-19. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kriz, A.J.; Sharp, P.A. Target specificity of the CRISPR-Cas9 system. Quant. Biol. 2014, 2, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Cui, B.; Kiga, K.; Aiba, Y.; Tan, X.-E.; Sato’o, Y.; Kawauchi, M.; Boonsiri, T.; Thitiananpakorn, K.; Taki, Y. Composition and diversity of CRISPR-Cas13a systems in the genus Leptotrichia. Front. Microbiol. 2019, 10, 2838. [Google Scholar] [CrossRef]
- Wan, P.; Cui, S.; Ma, Z.; Chen, L.; Li, X.; Zhao, R.; Xiong, W.; Zeng, Z. Reversal of mcr-1-mediated colistin resistance in Escherichia coli by CRISPR-Cas9 system. Infect. Drug Resist. 2020, 13, 1171–1178. [Google Scholar] [CrossRef]
- Rodrigues, M.; McBride, S.W.; Hullahalli, K.; Palmer, K.L.; Duerkop, B.A. Conjugative delivery of CRISPR-Cas9 for the selective depletion of antibiotic-resistant enterococci. Antimicrob. Agents Chemother. 2019, 63, e01454-19. [Google Scholar] [CrossRef]
- Wu, Y.; Battalapalli, D.; Hakeem, M.J.; Selamneni, V.; Zhang, P.; Draz, M.S.; Ruan, Z. Engineered CRISPR-Cas systems for the detection and control of antibiotic-resistant infections. J. Nanobiotechnol. 2021, 19, 401. [Google Scholar] [CrossRef]
- Ahmed, M.M.; Kayode, H.H.; Okesanya, O.J.; Ukoaka, B.M.; Eshun, G.; Mourid, M.R.; Adigun, O.A.; Ogaya, J.B.; Mohamed, Z.O.; Lucero-Prisno, D.E., 3rd. CRISPR-Cas Systems in the Fight Against Antimicrobial Resistance: Current Status, Potentials, and Future Directions. Infect. Drug Resist. 2024, 17, 5229–5245. [Google Scholar] [CrossRef]
- Jiang, J.; Cienfuegos-Gallet, A.V.; Long, T.; Peirano, G.; Chu, T.; Pitout, J.D.D.; Kreiswirth, B.N.; Chen, L. Intricate interplay of CRISPR-Cas systems, anti-CRISPR proteins, and antimicrobial resistance genes in a globally successful multi-drug resistant Klebsiella pneumoniae clone. Genome Med. 2025, 17, 9. [Google Scholar] [CrossRef]
- Tao, S.; Chen, H.; Li, N.; Liang, W. The application of the CRISPR-Cas system in antibiotic resistance. Infect. Drug Resist. 2022, 15, 4155–4168. [Google Scholar] [CrossRef]
- Panthi, V.K.; Fairfull-Smith, K.E.; Islam, N. Liposomal drug delivery strategies to eradicate bacterial biofilms: Challenges, recent advances, and future perspectives. Int. J. Pharm. 2024, 655, 124046. [Google Scholar] [CrossRef] [PubMed]
- Gholizadeh, P.; Aghazadeh, M.; Ghotaslou, R.; Rezaee, M.A.; Pirzadeh, T.; Cui, L.; Watanabe, S.; Feizi, H.; Kadkhoda, H.; Kafil, H.S. Role of CRISPR-Cas system on antibiotic resistance patterns of Enterococcus faecalis. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 49. [Google Scholar] [CrossRef] [PubMed]
- García-Gutiérrez, E.; Almendros, C.; Mojica, F.J.; Guzmán, N.M.; García-Martínez, J. CRISPR content correlates with the pathogenic potential of Escherichia coli. PLoS ONE 2015, 10, e0131935. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Draz, M.S.; Gu, M.; Yu, W.; Ruan, Z.; Luo, Q. Novel strategy to combat antibiotic resistance: A sight into the combination of CRISPR/Cas9 and nanoparticles. Pharmaceutics 2021, 13, 352. [Google Scholar] [CrossRef]
- Chen, Z.; Ma, L.; Bu, S.; Zhang, W.; Chen, J.; Li, Z.; Hao, Z.; Wan, J. CRISPR/Cas12a and immuno-RCA based electrochemical biosensor for detecting pathogenic bacteria. J. Electroanal. Chem. 2021, 901, 115755. [Google Scholar] [CrossRef]
- Wang, J.; Teng, Y.; Zhang, R.; Wu, Y.; Lou, L.; Zou, Y.; Li, M.; Xie, Z.-R.; Yan, Y. Engineering a PAM-flexible SpdCas9 variant as a universal gene repressor. Nat. Commun. 2021, 12, 6916. [Google Scholar] [CrossRef]
- Khan, I.; Saeed, K.; Khan, I. Nanoparticles: Properties, applications and toxicities. Arab. J. Chem. 2019, 12, 908–931. [Google Scholar] [CrossRef]
- Schairer, D.O.; Chouake, J.S.; Nosanchuk, J.D.; Friedman, A.J. The potential of nitric oxide releasing therapies as antimicrobial agents. Virulence 2012, 3, 271–279. [Google Scholar] [CrossRef]
- Blecher, K.; Nasir, A.; Friedman, A. The growing role of nanotechnology in combating infectious disease. Virulence 2011, 2, 395–401. [Google Scholar] [CrossRef]
- Knetsch, M.L.; Koole, L.H. New strategies in the development of antimicrobial coatings: The example of increasing usage of silver and silver nanoparticles. Polymers 2011, 3, 340–366. [Google Scholar] [CrossRef]
- Friedman, A.J.; Phan, J.; Schairer, D.O.; Champer, J.; Qin, M.; Pirouz, A.; Blecher-Paz, K.; Oren, A.; Liu, P.T.; Modlin, R.L. Antimicrobial and anti-inflammatory activity of chitosan–alginate nanoparticles: A targeted therapy for cutaneous pathogens. J. Investig. Dermatol. 2013, 133, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pornpattananangkul, D.; Hu, C.-M.; Huang, C.-M. Development of nanoparticles for antimicrobial drug delivery. Curr. Med. Chem. 2010, 17, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Holger Dana, J.; El Ghali, A.; Bhutani, N.; Lev Katherine, L.; Stamper, K.; Kebriaei, R.; Kunz Coyne Ashlan, J.; Morrisette, T.; Shah, R.; Alexander, J.; et al. Phage-antibiotic combinations against multidrug-resistant Pseudomonas aeruginosa in in vitro static and dynamic biofilm models. Antimicrob. Agents Chemother. 2023, 67, e00578-23. [Google Scholar] [CrossRef] [PubMed]
- Baptista, P.V.; McCusker, M.P.; Carvalho, A.; Ferreira, D.A.; Mohan, N.M.; Martins, M.; Fernandes, A.R. Nano-strategies to fight multidrug resistant bacteria—“A Battle of the Titans”. Front. Microbiol. 2018, 9, 1441. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Wang, P.; Hou, J.; Yao, Y.; Liu, Z.; Liu, S.; Li, T. Distinct community structure and microbial functions of biofilms colonizing microplastics. Sci. Total Environ. 2019, 650, 2395–2402. [Google Scholar] [CrossRef]
- Lee, N.-Y.; Ko, W.-C.; Hsueh, P.-R. Nanoparticles in the treatment of infections caused by multidrug-resistant organisms. Front. Pharmacol. 2019, 10, 1153. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, G.; Jin, S.; Xu, L.; Zhao, C.X. Development of High-Drug-Loading Nanoparticles. ChemPlusChem 2020, 85, 2143–2157. [Google Scholar] [CrossRef]
- Piktel, E.; Suprewicz, Ł.; Depciuch, J.; Chmielewska, S.; Skłodowski, K.; Daniluk, T.; Król, G.; Kołat-Brodecka, P.; Bijak, P.; Pajor-Świerzy, A. Varied-shaped gold nanoparticles with nanogram killing efficiency as potential antimicrobial surface coatings for the medical devices. Sci. Rep. 2021, 11, 12546. [Google Scholar] [CrossRef]
- Ali, S.G.; Ansari, M.A.; Alzohairy, M.A.; Alomary, M.N.; AlYahya, S.; Jalal, M.; Khan, H.M.; Asiri, S.M.M.; Ahmad, W.; Mahdi, A.A. Biogenic gold nanoparticles as potent antibacterial and antibiofilm nano-antibiotics against Pseudomonas aeruginosa. Antibiotics 2020, 9, 100. [Google Scholar] [CrossRef]
- Sguizzato, M.; Ferrara, F.; Baraldo, N.; Bondi, A.; Guarino, A.; Drechsler, M.; Valacchi, G.; Cortesi, R. Bilosomes and Biloparticles for the Delivery of Lipophilic Drugs: A Preliminary Study. Antioxidants 2023, 12, 2025. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Hemeg, H.A. Nanomaterials for alternative antibacterial therapy. Int. J. Nanomed. 2017, 12, 8211–8225. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Chen, Z.; Paul, P.K.; Lu, Y.; Wu, W.; Qi, J. Oral delivery of proteins and peptides: Challenges, status quo and future perspectives. Acta Pharm. Sin. B 2021, 11, 2416–2448. [Google Scholar] [CrossRef] [PubMed]
- Aslam, B.; Rasool, M.; Idris, A.; Muzammil, S.; Alvi, R.F.; Khurshid, M.; Rasool, M.H.; Zhang, D.; Ma, Z.; Baloch, Z. CRISPR-Cas system: A potential alternative tool to cope antibiotic resistance. Antimicrob. Resist. Infect. Control 2020, 9, 131. [Google Scholar] [CrossRef]
- Wei, J.; Peng, N.; Liang, Y.; Li, K.; Li, Y. Phage therapy: Consider the past, embrace the future. Appl. Sci. 2020, 10, 7654. [Google Scholar] [CrossRef]
- Mayegowda, S.B.; Ng, M.; Alghamdi, S.; Atwah, B.; Alhindi, Z.; Islam, F. Role of antimicrobial drug in the development of potential therapeutics. Evid. -Based Complement. Altern. Med. 2022, 2022, 2500613. [Google Scholar] [CrossRef]
- Olatunji, A.O.; Olaboye, J.A.; Maha, C.C.; Kolawole, T.O.; Abdul, S. Next-Generation strategies to combat antimicrobial resistance: Integrating genomics, CRISPR, and novel therapeutics for effective treatment. Eng. Sci. Technol. J. 2024, 5, 2284–2303. [Google Scholar] [CrossRef]

| Categories | Name | Mechanism | Function | Reference |
|---|---|---|---|---|
| Metallo-β-lactamases | blaVIM, blaIMP, and blaNDM | encode metallo-β-lactamases (MBLs), | confer resistance to carbapenems | [42] |
| Serine carbapenemases | blaOXA | encodes oxacillinases | confer resistance to carbapenems | [43] |
| Chromosomal β-Lactamase | ampC | overexpression of ampC, a chromosomal β-lactamase | hydrolyzes carbapenems, particularly when combined with oprD mutations. | [44] |
| pmrAB (PhoPQ) Two-Component System | pmrAB, phoPQ | lead to LPS modifications | decreasing colistin binding | [45] |
| Lipid A Biosynthesis Genes | lpxA, lpxC, and lpxD | lead to the complete loss of LPS production | resulting in colistin resistance through the absence of the drug’s primary target | [46] |
| Efflux Pump Genes | oprD, mexX, mexY | encode multidrug efflux pumps | expel a wide range of antibiotics | [47] |
| Aminoglycoside Resistance Genes | aac, aph, and aad | encode enzymes that modify aminoglycosides, rendering them ineffective | confer resistance to aminoglycoside | [48] |
| Quinolone Resistance Genes | qnr | encodes proteins that protect DNA gyrase and topoisomerase IV from quinolone inhibition, | confer resistance to quinolone | [49] |
| Key Plasmids Associated with AMR | pMOS94 | carries the blaVIM-1 gene within a class 1 integron (In70) | associated with the dissemination of metallo-β-lactamase genes among Pseudomonas species | [42] |
| pBT2436 and pBT2101 | carry extensive arrays of antibiotic resistance genes, including blaOXA-10, VEB-1, and aadA. | confer resistance to carbapenems, aminoglycoside and β-lactam | [50] | |
| pUM505 | encodes a mobile genetic element (Mpe) that confers resistance to heavy metals (e.g., chromate and mercury) and enhances virulence. | [51] | ||
| pAER57 | associated with the spread of blaVIM-2 | confer resistance to β-lactam | [42] | |
| pMKPA34-1 | carries resistance genes such as mexCD-oprJ and is associated with multidrug resistance | confer resistance to multiple antibiotics by inducing antibiotic efflux. | [50] |
| Peptide Name | Origin | Characteristics | Applications/Effects on P. aeruginosa | Reference |
|---|---|---|---|---|
| Colistin (Polymyxin E) | Bacillus polymyxa | Cationic, cyclic lipopeptide; targets lipopolysaccharides (LPS) in Gram-negative bacteria | Last-resort treatment for MDR/XDR P. aeruginosa; disrupts bacterial membrane integrity, leading to cell lysis. | [50] |
| LL-37 | Human cathelicidin | Cationic, α-helical peptide; part of the innate immune system | Broad-spectrum activity; disrupts P. aeruginosa biofilms and enhances immune cell recruitment. | [167] |
| Melittin | Honeybee venom | Cationic, α-helical peptide; highly amphipathic | Disrupts bacterial membranes; effective against P. aeruginosa biofilms and planktonic cells. | [168] |
| Pexiganan (MSI-78) | Synthetic analog of magainin | Cationic, α-helical peptide; derived from frog skin peptides | Targets P. aeruginosa membranes; used in topical treatments for wound infections. | [169] |
| Polymyxin B | Bacillus polymyxa | Cationic, cyclic lipopeptide; similar to colistin | Used against MDR/XDR P. aeruginosa; disrupts LPS and membrane integrity. | [170] |
| Lactoferrin | Mammalian secretions (e.g., milk, tears) | Iron-binding glycoprotein; cationic and multifunctional | Inhibits P. aeruginosa biofilm formation; enhances the activity of other antibiotics. | [171] |
| Cecropin A | Silk moth (Hyalophora cecropia) | Cationic, α-helical peptide; broad-spectrum activity | Disrupts P. aeruginosa membranes; effective against planktonic and biofilm-associated cells. | [172] |
| Defensins (e.g., HBD-1, HBD-2) | Human epithelial cells | Cationic, β-sheet peptides; part of the innate immune system | Disrupts P. aeruginosa membranes and biofilms; enhances immune responses. | [173] |
| Thanatin | Spined soldier bug (Podisus maculiventris) | Cationic, β-hairpin peptide; broad-spectrum activity | Targets P. aeruginosa membranes and inhibits outer membrane protein assembly. | [174] |
| Epidermin | Staphylococcus epidermidis | Lantibiotic; post-translationally modified peptide | Disrupts P. aeruginosa membranes; effective against biofilms and planktonic cells. | [175] |
| Plectasin | Fungus (Pseudoplectania nigrella) | Defensin-like peptide; cationic and stable | Disrupts P. aeruginosa membranes; effective against MDR strains. | [176] |
| Name | Source | Chemical Property | Inhibition Mechanism | Effect | References |
|---|---|---|---|---|---|
| Ajoene | Garlic (Allium sativum L.) | Natural product | Down-regulation of QS genes (lasA, chiC and rhlAB) | Inhibition of virulence and biofilm formation | [187,188] |
| Iberin | Horseradish extracts (Armoracia rusticana) | Natural product | Antagonists of LasIR and RhlI/R | Inhibition of virulence and biofilm formation | [189] |
| Sulforaphane | Broccoli extracts (Brassica oleracea) | Natural product | Antagonist of LasR | Inhibition of virulence and biofilm formation | [190] |
| Phenolics | Ginger extract (Curcuma longa) | Natural product | Down-regulation of LasI by binding of the compound’s long acyl chain to LasR | Inhibition of virulence and biofilm formation | [191] |
| Caffeine | Fenugreek seeds extract (Trigonella foenum-graecum L.) | Natural product | Inhibit AHL production | Inhibition of virulence and biofilm formation | [192] |
| Flavan-3-ol catechin | Malagasy plant (Combretum albiflorum) | Natural product | Reduced signal perception of RhlR | Inhibition of virulence and biofilm formation | [193] |
| Kalanchoeleaves extract | Kalanchoe blossfeldiana | Natural product | Interference with AHL production | Inhibition of virulence and biofilm formation | [194] |
| Clove oil | Syzygium aromaticum | Natural product | Inhibition of QS-mediated biofilm formation and disruption of already formed P. aeruginosa | Inhibition of virulence and biofilm formation | [195] |
| Juglone | Green part of Juglans regia | Natural product | bind to the PqsR active site | Inhibition of QS-mediated biofilms and reduction in virulence factor production | [196] |
| Quercetin (flavanols) | Apples, grapes, onions, tomatoes, etc. | Natural product | Reduced expression of the QS genes lasI, lasR, rhlI and rhlR | Significantly reduces biofilm formation | [197] |
| AHL-lactonase | Bacillus spp. gene aiiA | Natural product | Break down AHL | Prevents biofilm formation and reduces virulence factors of many bacteria | [198] |
| Paraoxonase | Human epithelial cells and serum from mammals such as rats, goats, cows and horses | Natural product | Inhibition of AHL-mediated QS in P. aeruginosa | Inhibition of virulence and biofilm formation | [199] |
| Vanillin | primary phenolic aldehyde of vanilla bean | Natural product | inhibited pqs expression and its associated phenotypes production | Inhibition of biofilm growth and reducing virulence | [200] |
| MAb RS2-1G9 | antibody | analog of the AHL acyl chain | Against the production of chlorpyrifosin in P. aeruginosa | [201] | |
| MAb XYD-11G2 | antibody | Hydrolysed 3-oxo-C12-HSL | Inhibition of pyocyanin production by P. aeruginosa | [201] | |
| Macrolides, including azithromycin and erythromycin | Synthetic product | Reducing transcription of lasI and rhlI simultaneously reduce the concentration of 3-oxo-C12-HSL and C4-HSL | Reducing the production of group induction-dependent virulence factors. | [202,203] | |
| Thiazolidinedione (TZD) and its derivatives | Synthetic product | - | A 70 percent reduction in biomass in biofilm | [204] | |
| Furanone compounds | Synthetic product | - | Inhibition of QS and reducing bacteria growth | [205] | |
| Cyclohexanone analog of HSL | Synthetic product | - | Effective antagonism of QS-mediated activity, including biofilm formation | [206] | |
| Engineered variant of hyper-thermostable lactonaseSsoPox | Sulfolobus solfataricus | Synthetic molecule | Degradation of the lactone ring of 3-oxo-C12 AHL and enhancement of catalytic efficiency | Reducing the severity of pneumonia caused by P. aeruginosa infection | [207] |
| Bacteria | Gene | Mechanism | Reference |
|---|---|---|---|
| C. difficile | RNase Y | Using bacteriophage ϕCD24-2 expressing bacterial genome-targeting crRNAs/chromosomal DNA degradation | [224] |
| K. pneumoniae | ramR, tetA, mgrB | pSGKP-spe and pBECKP-spe plasmids engineered with the CRISPR-Cas9 system | [226] |
| S. aureus | nuc, esxA | Using Mild Phage ϕSaBov as chromosomal DNA degradation delivery | [222] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, M.; Chua, S.L. Antibiotic-Resistant Pseudomonas aeruginosa: Current Challenges and Emerging Alternative Therapies. Microorganisms 2025, 13, 913. https://doi.org/10.3390/microorganisms13040913
Hu M, Chua SL. Antibiotic-Resistant Pseudomonas aeruginosa: Current Challenges and Emerging Alternative Therapies. Microorganisms. 2025; 13(4):913. https://doi.org/10.3390/microorganisms13040913
Chicago/Turabian StyleHu, Minqi, and Song Lin Chua. 2025. "Antibiotic-Resistant Pseudomonas aeruginosa: Current Challenges and Emerging Alternative Therapies" Microorganisms 13, no. 4: 913. https://doi.org/10.3390/microorganisms13040913
APA StyleHu, M., & Chua, S. L. (2025). Antibiotic-Resistant Pseudomonas aeruginosa: Current Challenges and Emerging Alternative Therapies. Microorganisms, 13(4), 913. https://doi.org/10.3390/microorganisms13040913