Genetic Traces of the Francisella tularensis Colonization of Spain, 1998–2020

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains
2.2. Genome Sequencing
2.3. Taxonomic Classification
2.4. Sizing of PCR Products Targeting VNTRs with Capillary Electrophoresis and Genome Sequencing
2.5. Phylogenetics and Phylogeography
3. Results
3.1. Genome Sequencing, Assembly and SNP Identification
3.2. Taxonomic Classification
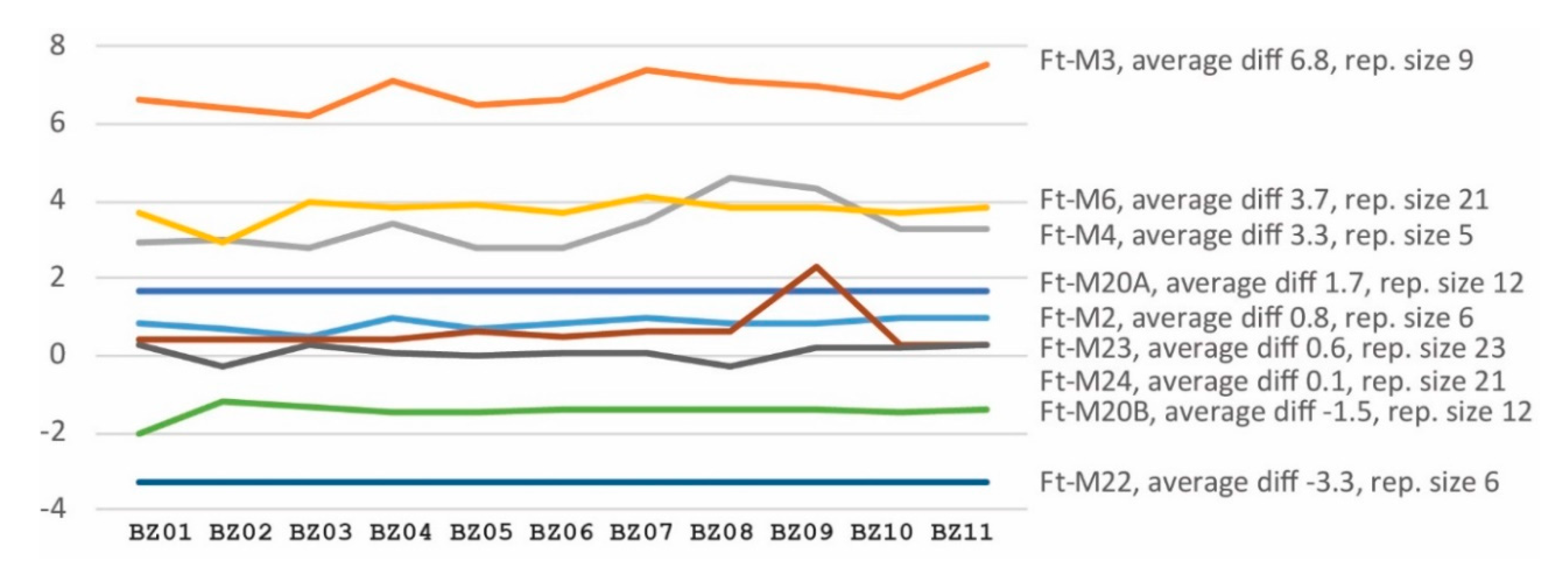
3.3. Analysis of VNTRs by Short Read Genome Sequencing and Capillary Electrophoresis
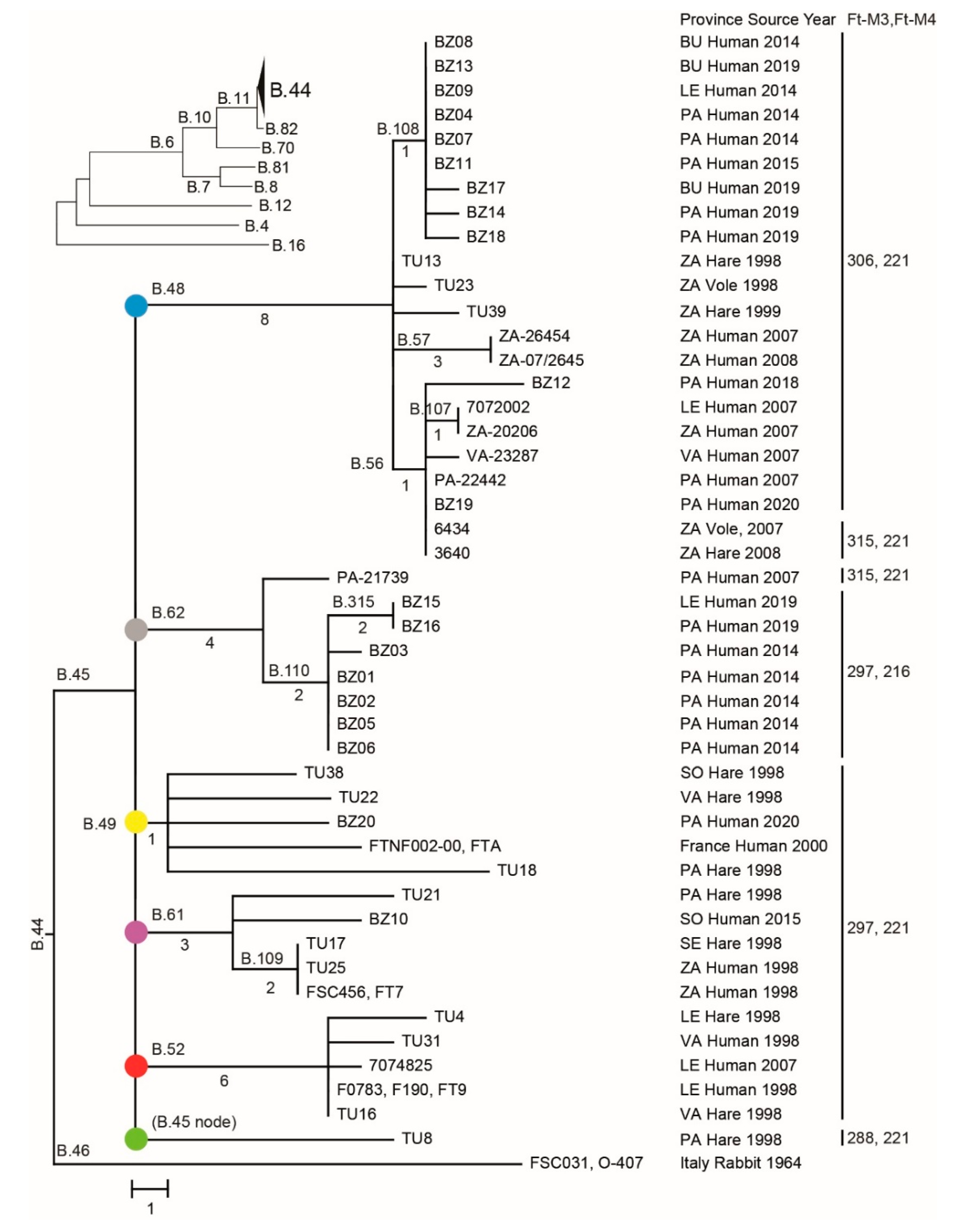
3.4. Phylogenetic Analysis Using SNP or VNTR Data
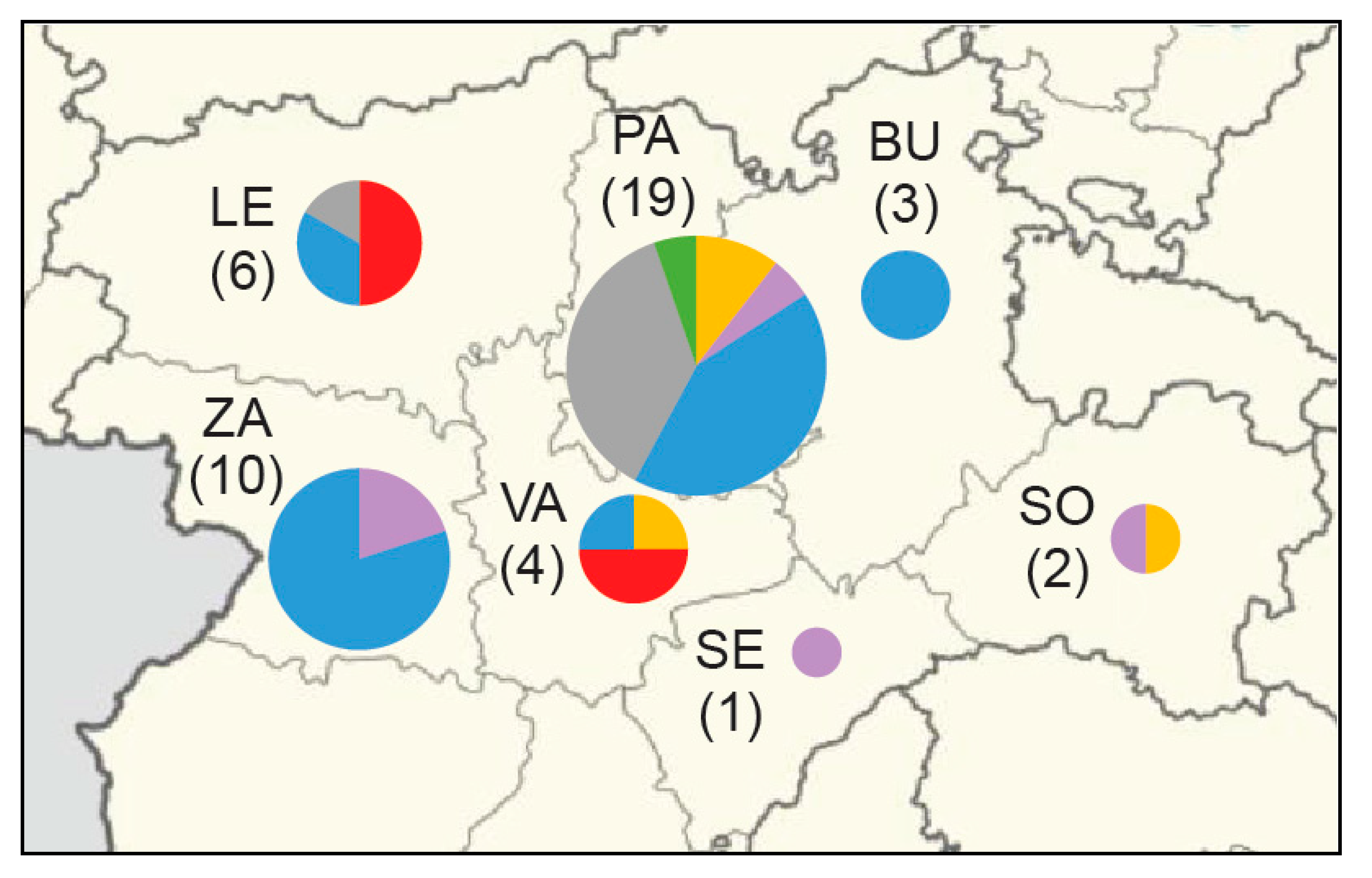
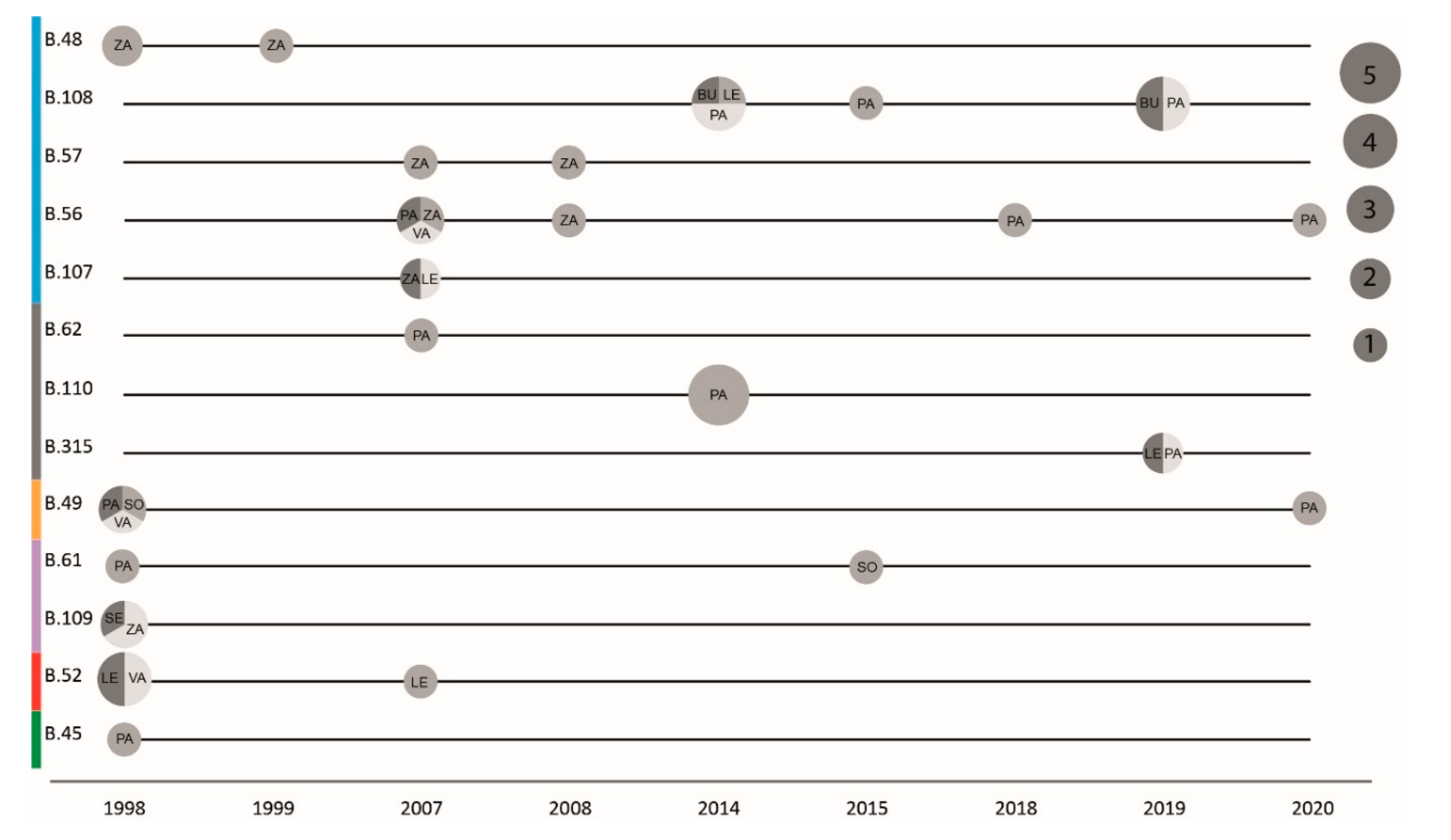
3.5. Phylogeography
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Source | Province | Year of Isolation | Accession 1 | Clade |
|---|---|---|---|---|---|
| F0783, FT9, F190 | Human | León | 1998 | SAMN03773734 | B.52 |
| TU4 | Hare | León | 1998 | SAMN03773884 | B.52 |
| TU21 | Hare | Palencia | 1998 | SAMN03773885 | B.61 |
| TU39 | Hare | Zamora | 1999 | SAMN03773886 | B.48 |
| TU8 | Hare | Palencia | 1998 | SAMN03773887 | B.45 |
| TU17 | Hare | Segovia | 1998 | SAMN03773888 | B.61 |
| TU18 | Hare | Palencia | 1998 | SAMN03773889 | B.49 |
| TU16 | Hare | Valladolid | 1998 | SAMN03773890 | B.52 |
| TU38 | Hare | Soria | 1998 | SAMN03773891 | B.49 |
| TU23 | Vole | Zamora | 1998 | SAMN03773892 | B.48 |
| TU22 | Hare | Valladolid | 1998 | SAMN03773893 | B.49 |
| TU31 | Human | Valladolid | 1998 | SAMN03773894 | B.52 |
| TU13 | Hare | Zamora | 1998 | SAMN03773895 | B.48 |
| TU25 | Human | Zamora | 1998 | SAMN03773896 | B.61 |
| VA-23287 | Human | Valladolid | 2007 | SAMN03773897 | B.56 |
| ZA-07/2645 | Human | Zamora | 2008 | SAMN03773898 | B.57 |
| 7072002 | Human | León | 2007 | SAMN03773899 | B.56 |
| 6434 | Vole | Zamora | 2007 | SAMN03773900 | B.56 |
| 3640 | Hare | Zamora | 2008 | SAMN03773901 | B.56 |
| ZA-26454 | Human | Zamora | 2007 | SAMN03773902 | B.57 |
| PA-21739 | Human | Palencia | 2007 | SAMN03773903 | B.62 |
| ZA-20206 | Human | Zamora | 2007 | SAMN03773904 | B.56 |
| 7074825 | Human | León | 2007 | SAMN03773905 | B.52 |
| PA-22442 | Human | Palencia | 2007 | SAMN03773906 | B.56 |
| FSC456, FT7 | Human | Zamora | 1998 | SAMN03774111 | B.61 |
| Primer | Sequence | Fluorescence Dye | Melting Point |
|---|---|---|---|
| Ft-M03_F | gcacgcttgtctcctatcatcctctggtagtc | HEX | 64.7 °C |
| Ft-M03_R | gaacaacaaaagcaacagcaaaattcacaaaa | 59.4 °C | |
| Ft-M04_F | gcgcgctatctaactaatttttatattgaaacaatcaaat | FAM | 59.3 °C |
| Ft-M04_R | gcaaatataccgtaatgccacctatgaaaactc | 60.2 °C | |
| Ft-M05_F | gtttgttacgccaataaacaaaaagtgtaaataatg | NED | 57.6 °C |
| Ft-M05_R | gctcagctcgaactccgtcataccttcttc | 64.1 °C | |
| Ft-M10_F | gctaattttttcatatttatctccatttttacttttttgc | HEX | 57 °C |
| Ft-M10_R | gctcagctcgaactccgtcataccttcttc | 64.1 °C | |
| Ft-M20A_F | gtatatcttggaataagccggagttagatggttct | FAM | 61.1 °C |
| FtM20A_R | gcaataactttatcacccttattgtagactgcttctgc | 62.1 °C | |
| Ft-M20B_F | gggtgataaagttattgttaatggtgtgacttatgaa | 59.5 °C | |
| Ft-M20B_R | gtaactacttgaccgccagtatatgcttgacct | HEX | 63 °C |
| Ft-M06_F | gtttttggtgaactgccaacaccataactt | NED | 61.1 °C |
| Ft-M06_R | gcaattcagcgaaaccctatcttagcctc | 62 °C | |
| Ft-M02_F | gctgctgtggctgtaaatgttgctatgct | FAM | 64.1 °C |
| Ft-M02_R | gcagggcacaattcttgaccatcagg | 63.3 °C | |
| Ft-M22_F | gtggaaatgcaaaaacaatatcgaggaacttta | FAM | 59.1 °C |
| Ft-M22_R | gttttttctcgtccgctgttagtgattttacatc | 60.5 °C | |
| Ft-M23_F | gctggattattaaagcatatgacagacgagtagg | NED | 60.6 °C |
| Ft-M23_R | gttccctcaggtttatccaaagttttaatgttttatt | 59 °C | |
| Ft-M24_F | gaatcttaatccatacggtcctaataatattcctgtcaat | NED | 59.8 °C |
| Ft-M24_R | gttggtacttatgggctatagcggatattatttcagt | 61.2 °C |
| ID | Total No. Bases | Total No. Contigs | N50 | Mean Sequence Depth | BioSample Accession * | Clade | Subclade Path |
|---|---|---|---|---|---|---|---|
| BZ01 | 1,810,591 | 95 | 27699 | 455 | SAMN12071621 | B.62 | B.62–B.110 |
| BZ02 | 1,821,580 | 95 | 27699 | 431 | SAMN12071622 | B.62 | B.62–B.110 |
| BZ03 | 1,825,760 | 111 | 23863 | 466 | SAMN12071623 | B.62 | B.62–B.110 |
| BZ04 | 1,823,438 | 98 | 26961 | 481 | SAMN12071624 | B.48 | B.48–B.108 |
| BZ05 | 1,809,217 | 97 | 25525 | 410 | SAMN12071625 | B.62 | B.62–B.110 |
| BZ06 | 1,825,942 | 105 | 25544 | 574 | SAMN12071626 | B.62 | B.62–B.110 |
| BZ07 | 1,812,133 | 98 | 26732 | 550 | SAMN12071627 | B.48 | B.48–B.108 |
| BZ08 | 1,818,495 | 94 | 27699 | 559 | SAMN12071628 | B.48 | B.48–B.108 |
| BZ09 | 1,825,893 | 108 | 25990 | 424 | SAMN12071629 | B.48 | B.48–B.108 |
| BZ10 | 1,811,236 | 88 | 33271 | 417 | SAMN12071630 | B.61 | B.61 |
| BZ11 | 1,808,131 | 92 | 29735 | 514 | SAMN12071631 | B.48 | B.48–B.108 |
| BZ12 | 1,830,400 | 100 | 26972 | 340 | SAMN16515973 | B.48 | B.48–B.56 |
| BZ13 | 1,756,606 | 218 | 15177 | 269 | SAMN16515974 | B.48 | B.48–B.108 |
| BZ14 | 1,775,418 | 201 | 15465 | 282 | SAMN16515975 | B.48 | B.48–B.108 |
| BZ15 | 1,818,738 | 99 | 26747 | 242 | SAMN16515976 | B.62 | B.62–B.110–B.315 |
| BZ16 | 1,813,941 | 97 | 26857 | 281 | SAMN16515977 | B.62 | B.62–B.110–B.315 |
| BZ17 | 1,819,785 | 97 | 27089 | 331 | SAMN16515978 | B.48 | B.48–B.108 |
| BZ18 | 1,708,033 | 369 | 7369 | 231 | SAMN16515979 | B.48 | B.48–B.108 |
| BZ19 | 1,833,295 | 94 | 27956 | 103 | SAMN16515980 | B.48 | B.48–B.56 |
| BZ20 | 1,834,431 | 89 | 29599 | 89 | SAMN16515981 | B.49 | B.49 |
| Clade | Genomes | No. SNPs | SNP Position (Anc/Der) |
|---|---|---|---|
| B.48 | BZ04:BZ07:BZ08:BZ09:BZ11: BZ12:BZ13:BZ14:BZ17:BZ18: BZ19:TU39:TU23:TU13: VA-23287:ZA-07/2645:7072002: 6434:3640:ZA-26454: ZA-20206:PA-22442 | 8 | 170,704 (C/T);258,998 (G/T); 736,278 (G/A);1,075,715 (C/T); 1,194,117 (G/A);1,225,490 (G/A); 1,558,703 (C/T);1,746,518 (C/T) |
| B.108 | BZ04:BZ07:BZ08:BZ09:BZ11: BZ13:BZ14:BZ17:BZ18 | 1 | 1,327,297 (C/T) |
| B.57 | ZA-07/2645:ZA-26454 | 3 | 668,630 (T/C);1,747,062 (G/T);1,860,651 (G/A) |
| B.56 | BZ12:BZ19:VA-23287: 7072002:6434:3640:ZA-20206: PA-22442 | 1 | 827,994 (G/A) |
| B.107 | 7072002:ZA-20206 | 1 | 520,520 (C/T) |
| B.62 | BZ01:BZ02:BZ03:BZ05:BZ06: BZ15:BZ16:PA-21739 | 4 | 155,417 (G/A);411,903 (C/T); 505,602 (C/T);691,772 (C/T) |
| B.110 | BZ01:BZ02:BZ03:BZ05:BZ06: BZ15:BZ16 | 2 | 1,301,561 (G/A);1,742,563 (G/A) |
| B.315 | BZ15:BZ16 | 2 | 975,621 (G/C);1,721,277 (T/C) |
| B.61 | BZ10:TU21:TU17:TU25:FSC456 | 3 | 235,472 (G/A);1,009,840 (G/T);1,332,119 (G/A) |
| B.109 | TU17:TU25:FSC456 | 2 | 1,082,513 (T/C);1,171,726 (G/A) |
| B.49 | BZ20:TU18:TU38:TU22:FTNF002 | 1 | 224,367 (G/A) |
| B.52 | F0783:TU4:TU16:TU31:7074825 | 6 | 238,894 (C/T);324,043 (C/T); 1,009,935 (C/T);1,130,906 (C/T); 1,151,666 (T/C);444,395 (C/T) |
| ID | Ft-M2 | Ft-M3 | Ft-M4 | Ft-M5 | Ft-M6 | Ft-M10 | Ft-M20A | Ft-M20B | Ft-M22 | Ft-M23 | Ft-M24 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BZ01 | 339.8 | 303.6 | 218.9–219.9 | 299.6 | 275.7 | 199.6 | 307.7 | 151.0 | 174.7 | 327.4 | 400.3 |
| BZ02 | 339.7 | 303.4 | 219.0 | 300.0 | 274.9 | 199.6 | 307.7 | 151.8 | 174.7 | 327.4 | 399.7 |
| BZ03 | 339.5 | 303.2 | 218.8–219.8 | 300.3 | 276.0 | 199.6 | 307.7 | 151.7 | 174.7 | 327.4 | 400.3 |
| BZ04 | 340.0 | 313.1 | 224.4 | 300.3 | 275.8 | 199.6 | 307.7 | 151.5 | 174.7 | 327.4 | 400.1 |
| BZ05 | 339.7 | 303.5 | 218.8–219.8 | 300.0 | 275.9 | 199.6 | 307.7 | 151.5 | 174.7 | 327.6 | 400.0 |
| BZ06 | 339.8 | 303.6 | 218.8–219.8 | 300.1 | 275.7 | 199.6 | 307.7 | 151.6 | 174.7 | 327.5 | 400.1 |
| BZ07 | 340.0 | 313.4 | 224.5 | 300.3 | 276.1 | 199.6 | 307.7 | 151.6 | 174.7 | 327.6 | 400.1 |
| BZ08 | 339.8 | 313.1 | 225.6 | 299.7 | 275.8 | 199.6 | 307.7 | 151.6 | 174.7 | 327.6 | 399.7 |
| BZ09 | 339.8 | 313.0 | 225.3 | 300.2 | 275.8 | 199.6 | 307.7 | 151.6 | 174.7 | 327.3 | 400.2 |
| BZ10 | 340.0 | 303.7 | 224.3 | 300.2 | 275.7 | 199.6 | 307.7 | 151.5 | 174.7 | 327.3 | 400.2 |
| BZ11 | 340.0 | 313.5 | 224.3 | 300.1 | 275.8 | 199.6 | 307.7 | 151.6 | 174.7 | 327.3 | 400.3 |
| ID | Ft-M2 | Ft-M3 | Ft-M4 | Ft-M5 | Ft-M6 | Ft-M10 | Ft-M20A | Ft-M20B | Ft-M22 | Ft-M23 | Ft-M24 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BZ01 | 339 | 297 | 216 | - | 272 | - | 306 | 153 | 178 | 327 | 400 |
| BZ02 | 339 | 297 | 216 | - | 272 | - | 306 | 153 | 178 | 327 | 400 |
| BZ03 | 339 | 297 | 216 | 298 | 272 | - | 306 | 153 | 178 | 327 | 400 |
| BZ04 | 339 | 306 | 221 | 298 | 272 | 197 | 306 | 153 | 178 | 327 | 400 |
| BZ05 | 339 | 297 | 216 | - | 272 | - | 306 | 153 | 178 | 327 | 400 |
| BZ06 | 339 | 297 | 216 | 298 | 272 | 197 | 306 | 153 | 178 | 327 | 400 |
| BZ07 | 339 | 306 | 221 | - | 272 | - | 306 | 153 | 178 | 327 | 400 |
| BZ08 | 339 | 306 | 221 | 289 * | 272 | 188 * | 306 | 153 | 178 | 327 | 400 |
| BZ09 | 339 | 306 | 221 | - | 272 | - | 306 | 153 | 178 | 325 | 400 |
| BZ10 | 339 | 297 | 221 | 298 | 272 | 197 | 306 | 153 | 178 | 327 | 400 |
| BZ11 | 339 | 306 | 221 | - | 272 | - | 306 | 153 | 178 | 327 | 400 |
| ID | Ft-M2 | Ft-M3 | Ft-M4 | Ft-M6 | Ft-M20A | Ft-M20B | Ft-M22 | Ft-M23 | Ft-M24 |
|---|---|---|---|---|---|---|---|---|---|
| BZ01 | 0.8 | 6.6 | 2.9 | 3.7 | 1.7 | −2 | −3.3 | 0.4 | 0.3 |
| BZ02 | 0.7 | 6.4 | 3 | 2.9 | 1.7 | −1.2 | −3.3 | 0.4 | −0.3 |
| BZ03 | 0.5 | 6.2 | 2.8 | 4 | 1.7 | −1.3 | −3.3 | 0.4 | 0.3 |
| BZ04 | 1 | 7.1 | 3.4 | 3.8 | 1.7 | −1.5 | −3.3 | 0.4 | 0.1 |
| BZ05 | 0.7 | 6.5 | 2.8 | 3.9 | 1.7 | −1.5 | −3.3 | 0.6 | 0 |
| BZ06 | 0.8 | 6.6 | 2.8 | 3.7 | 1.7 | −1.4 | −3.3 | 0.5 | 0.1 |
| BZ07 | 1 | 7.4 | 3.5 | 4.1 | 1.7 | −1.4 | −3.3 | 0.6 | 0.1 |
| BZ08 | 0.8 | 7.1 | 4.6 | 3.8 | 1.7 | −1.4 | −3.3 | 0.6 | −0.3 |
| BZ09 | 0.8 | 7 | 4.3 | 3.8 | 1.7 | −1.4 | −3.3 | 2.3 | 0.2 |
| BZ10 | 1 | 6.7 | 3.3 | 3.7 | 1.7 | −1.5 | −3.3 | 0.3 | 0.2 |
| BZ11 | 1 | 7.5 | 3.3 | 3.8 | 1.7 | −1.4 | −3.3 | 0.3 | 0.3 |
| Median diff. | 0.8 | 6.7 | 3.3 | 3.8 | 1.7 | −1.4 | −3.3 | 0.4 | 0.1 |
| Average diff. | 0.8 | 6.8 | 3.3 | 3.7 | 1.7 | −1.5 | −3.3 | 0.6 | 0.1 |
| Repeat size | 6 | 9 | 5 | 21 | 12 | 12 | 6 | 23 | 21 |
References
- Keim, P.; Johansson, A.; Wagner, D.M.; Epidemiology, M. Molecular epidemiology, evolution, and ecology of Francisella. Ann. N. Y. Acad. Sci. 2007, 1105, 30–66. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.R.; Steinhaus, E.A.; Kohls, G.M.; Jellison, W.L. Contamination of natural waters and mud with Pasteurella tularensis and tularemia in beavers and muskrats in the northwestern United States. Bull. Natl. Inst. Health 1951, 193, 1–161. [Google Scholar] [PubMed]
- Broman, T.; Thelaus, J.; Andersson, A.-C.; Bäckman, S.; Wikström, P.; Larsson, E.; Granberg, M.; Karlsson, L.; Bäck, E.; Eliasson, H.; et al. Molecular Detection of Persistent Francisella tularensis Subspecies holarctica in Natural Waters. Int. J. Microbiol. 2011, 2011, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, C.; Heuner, K.; Myrtennäs, K.; Karlsson, E.; Jacobs, D.; Kutzer, P.; Große, K.; Forsman, M.; Grunow, R. High and novel genetic diversity of Francisella tularensis in Germany and indication of environmental persistence. Epidemiol. Infect. 2016, 144, 3025–3036. [Google Scholar] [CrossRef] [Green Version]
- Janse, I.; Maas, M.; Rijks, J.M.; Koene, M.; van der Plaats, R.Q.; Engelsma, M.; van der Tas, P.; Braks, M.; Stroo, A.; Notermans, D.W.; et al. Environmental surveillance during an outbreak of tularaemia in hares, the Netherlands, 2015. Eurosurveillance 2017, 22, 30607. [Google Scholar] [CrossRef] [Green Version]
- Janse, I.; van der Plaats, R.Q.J.; Van Passel, M.W.J. Environmental surveillance of zoonotic Francisella tularensis in the Netherlands. Front. Cell. Infect. Microbiol. 2018, 8, 140. [Google Scholar] [CrossRef]
- Vogler, A.J.; Birdsell, D.; Price, L.B.; Bowers, J.R.; Beckstrom-Sternberg, S.M.; Auerbach, R.K.; Beckstrom-Sternberg, J.S.; Johansson, A.; Clare, A.; Buchhagen, J.L.; et al. Phylogeography of Francisella tularensis: Global expansion of a highly fit clone. J. Bacteriol. 2009, 191, 2474–2484. [Google Scholar] [CrossRef] [Green Version]
- Svensson, K.; Granberg, M.; Karlsson, L.; Neubauerova, V.; Forsman, M.; Johansson, A. A real-time PCR array for hierarchical identification of Francisella isolates. PLoS ONE 2009, 4, e8360. [Google Scholar] [CrossRef]
- Svensson, K.; Bäck, E.; Eliasson, H.; Berglund, L.; Granberg, M.; Karlsson, L.; Larsson, P.; Forsman, M.; Johansson, A. Landscape Epidemiology of Tularemia Outbreaks in Sweden. Emerg. Infect. Dis. 2009, 15, 1937–1947. [Google Scholar] [CrossRef] [Green Version]
- Chanturia, G.; Birdsell, D.N.; Kekelidze, M.; Zhgenti, E.; Babuadze, G.; Tsertsvadze, N.; Tsanava, S.; Imnadze, P.; Beckstrom-Sternberg, S.M.; Beckstrom-Sternberg, J.S.; et al. Phylogeography of Francisella tularensis subspecies holarctica from the country of Georgia. BMC Microbiol. 2011, 11, 139. [Google Scholar] [CrossRef] [Green Version]
- Gyuranecz, M.; Birdsell, D.N.; Splettstoesser, W.; Seibold, E.; Beckstrom-sternberg, S.M.; Makrai, L.; Fodor, L.; Fabbi, M.; Vicari, N.; Johansson, A.; et al. Phylogeography of Francisella tularensis subsp. holarctica, Europe. Emerg. Infect. Dis. 2012, 18, 290–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, E.; Svensson, K.; Lindgren, P.; Byström, M.; Sjödin, A.; Forsman, M.; Johansson, A.; Karlsson, E.; Svensson, K.; Lindgren, P.; et al. The phylogeographic pattern of Francisella tularensis in Sweden indicates a Scandinavian origin of Eurosiberian tularaemia. Environ. Microbiol. 2013, 15, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Farlow, J.; Larsson, P.; Dukerich, M.; Chambers, E.; Bystrom, M.; Fox, J.; Chu, M.; Forsman, M.; Sjostedt, A.; et al. Worldwide Genetic Relationships among Francisella tularensis Isolates Determined by Multiple-Locus Variable-Number Tandem Repeat Analysis. J. Bacteriol. 2004, 186, 5808–5818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, A.; Forsman, M.; Sjöstedt, A. The development of tools for diagnosis of tularemia and typing of Francisella tularensis. APMIS 2004, 112, 898–907. [Google Scholar] [CrossRef]
- Timofeev, V.; Bakhteeva, I.; Titareva, G.; Kopylov, P.; Christiany, D.; Mokrievich, A.; Dyatlov, I.; Vergnaud, G. Russian isolates enlarge the known geographic diversity of Francisella tularensis subsp. mediasiatica. PLoS ONE 2017, 12, e0183714. [Google Scholar] [CrossRef] [Green Version]
- Dwibedi, C.; Birdsell, D.; Lärkeryd, A.; Myrtennäs, K.; Öhrman, C.; Nilsson, E.; Karlsson, E.; Hochhalter, C.; Rivera, A.; Maltinsky, S.; et al. Long-range dispersal moved Francisella tularensis into Western Europe from the East. Microb. Genom. 2016, 2. [Google Scholar] [CrossRef]
- Eiros Bouza, J.M.; Rodríguez-Torres, A. Tularemia. Rev. Clin. Esp. 1998, 198, 785–788. [Google Scholar]
- Farlow, J.; Smith, K.L.K.L.; Wong, J.; Abrams, M.; Lytle, M.; Keim, P. Francisella tularensis strain typing using multiple-locus, variable-number tandem repeat analysis. J. Clin. Microbiol. 2001, 39, 3186–3192. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.S.; Shan, G.; Kristiansen, K.; et al. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [Green Version]
- Lärkeryd, A.; Myrtennäs, K.; Karlsson, E.; Dwibedi, C.K.; Forsman, M.; Larsson, P.; Johansson, A.; Sjödin, A. CanSNPer: A hierarchical genotype classifier of clonal pathogens. Bioinformatics 2014, 30, 1762–1764. [Google Scholar] [CrossRef] [Green Version]
- Kreizinger, Z. Comparative Characterisation of Members of the Family Francisellaceae. Ph.D. Thesis, Szent István University, Doctoral School of Veterinary Science, Gödöllo, Hungary, 2016. [Google Scholar]
- Wittwer, M.; Altpeter, E.; Pilo, P.; Gygli, S.M.; Beuret, C.; Foucault, F.; Ackermann-Gäumann, R.; Karrer, U.; Jacob, D.; Grunow, R.; et al. Population Genomics of Francisella tularensis subsp. holarctica and its Implication on the Eco-Epidemiology of Tularemia in Switzerland. Front. Cell. Infect. Microbiol. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Appelt, S.; Köppen, K.; Radonić, A.; Drechsel, O.; Jacob, D.; Grunow, R.; Heuner, K. Genetic Diversity and Spatial Segregation of Francisella tularensis Subspecies holarctica in Germany. Front. Cell. Infect. Microbiol. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiwald, S.; Simeon, A.; Hofer, E.; Weiss, G.; Bellmann-Weiler, R. Tularemia Goes West: Epidemiology of an Emerging Infection in Austria. Microorganisms 2020, 8, 1597. [Google Scholar] [CrossRef] [PubMed]
- Kevin, M.; Girault, G.; Caspar, Y.; Cherfa, M.A.; Mendy, C.; Tomaso, H.; Gavier-Widen, D.; Escudero, R.; Maurin, M.; Durand, B.; et al. Phylogeography and Genetic Diversity of Francisella tularensis subsp. holarctica in France (1947–2018). Front. Microbiol. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões, M.; Breitkreuz, L.; Alvarado, M.; Baca, S.; Cooper, J.C.; Heins, L.; Herzog, K.; Lieberman, B.S. The Evolving Theory of Evolutionary Radiations. Trends Ecol. Evol. 2016, 31, 27–34. [Google Scholar] [CrossRef]
- Anda, P.; Segura del Pozo, J.; Díaz García, J.M.; Escudero, R.; García Peña, F.J.; López Velasco, M.C.; Sellek, R.E.; Jiménez Chillarón, M.R.; Sánchez Serrano, L.P.; Martínez Navarro, J.F. Waterborne outbreak of tularemia associated with crayfish fishing. Emerg. Infect. Dis. 2001, 7, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Allue, M.; Sopeña, C.R.; Gallardo, M.T.; Mateos, L.; Vian, E.; Garcia, M.J.; Ramos, J.; Berjon, A.C.; Viña, M.C.; Garcia, M.P.; et al. Tularaemia outbreak in Castilla y León, Spain, 2007: An update. Eurosurveillance 2008, 13, 18948. [Google Scholar] [CrossRef] [PubMed]
- Mörner, T. The ecology of tularaemia. Rev. Sci. Tech. 1992, 11, 1123–1130. [Google Scholar] [CrossRef] [Green Version]
- Luque-Larena, J.J.; Mougeot, F.; Arroyo, B.; Vidal, M.D.; Rodríguez-Pastor, R.; Escudero, R.; Anda, P.; Lambin, X. Irruptive mammal host populations shape tularemia epidemiology. PLoS Pathog. 2017, 13, 1–5. [Google Scholar] [CrossRef] [Green Version]




| ID | Source | Transmission Related to | Year | Province |
|---|---|---|---|---|
| BZ01 | Skin ulcer | Crayfish | 2014 | Palencia |
| BZ02 | Skin ulcer | Crayfish | 2014 | Palencia |
| BZ03 | Skin ulcer | Crayfish/rodents/gardening | 2014 | Palencia |
| BZ04 | Skin ulcer | Crayfish/countryside walks | 2014 | Palencia |
| BZ05 | Blood | Rodent/agriculture | 2014 | Palencia |
| BZ06 | Skin ulcer | Crayfish | 2014 | Palencia |
| BZ07 | Blood | Country house | 2014 | Palencia |
| BZ08 | Transbronchial needle aspiration of lymph node | Environment | 2014 | Burgos |
| BZ09 | Skin ulcer | Hunting | 2014 | Leon |
| BZ10 | Skin ulcer | Hunting | 2015 | Soria |
| BZ11 | Blood | Not available | 2015 | Palencia |
| BZ12 | Blood | Farming | 2018 | Palencia |
| BZ13 | Skin abscess | Crayfish | 2019 | Burgos |
| BZ14 | Blood | Gardening | 2019 | Palencia |
| BZ15 | Lymph node abscess | Not available | 2019 | León |
| BZ16 | Lymph node abscess | Crayfish | 2019 | Palencia |
| BZ17 | Skin ulcer | Crayfish | 2019 | Burgos |
| BZ18 | Skin ulcer | Hunting | 2019 | Palencia |
| BZ19 | Blood | Not available | 2020 | Palencia |
| BZ20 | Lymph node abscess | Countryside walks | 2020 | Palencia |
| Ft-M3 (9 bp Repeat Size) | Ft-M4 (5 bp Repeat Size) | |||||
|---|---|---|---|---|---|---|
| ID | No. Repeats | Assay | WGS | No. Repeats | Assay | WGS |
| BZ01 | 8 | 303.6 | 297 | 1 | 218.9–219.9 | 216 |
| BZ02 | 8 | 303.4 | 297 | 1 | 219.0 | 216 |
| BZ03 | 8 | 303.2 | 297 | 1 | 218.8–219.8 | 216 |
| BZ04 | 9 | 313.1 | 306 | 2 | 224.4 | 221 |
| BZ05 | 8 | 303.5 | 297 | 1 | 218.8–219.8 | 216 |
| BZ06 | 8 | 303.6 | 297 | 1 | 218.8–219.8 | 216 |
| BZ07 | 9 | 313.4 | 306 | 2 | 224.5 | 221 |
| BZ08 | 9 | 313.1 | 306 | 2 | 225.6 | 221 |
| BZ09 | 9 | 313.0 | 306 | 2 | 225.3 | 221 |
| BZ10 | 8 | 303.7 | 297 | 2 | 224.3 | 221 |
| BZ11 | 9 | 313.5 | 306 | 2 | 224.3 | 221 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Myrtennäs, K.; Escudero, R.; Zaballos, Á.; González-Martín-Niño, R.; Gyuranecz, M.; Johansson, A. Genetic Traces of the Francisella tularensis Colonization of Spain, 1998–2020. Microorganisms 2020, 8, 1784. https://doi.org/10.3390/microorganisms8111784
Myrtennäs K, Escudero R, Zaballos Á, González-Martín-Niño R, Gyuranecz M, Johansson A. Genetic Traces of the Francisella tularensis Colonization of Spain, 1998–2020. Microorganisms. 2020; 8(11):1784. https://doi.org/10.3390/microorganisms8111784
Chicago/Turabian StyleMyrtennäs, Kerstin, Raquel Escudero, Ángel Zaballos, Rosa González-Martín-Niño, Miklós Gyuranecz, and Anders Johansson. 2020. "Genetic Traces of the Francisella tularensis Colonization of Spain, 1998–2020" Microorganisms 8, no. 11: 1784. https://doi.org/10.3390/microorganisms8111784