
Full-Length Genome and Partial Viral Genes Phylogenetic and Geographical Analysis of Dengue Serotype 3 Isolates

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Analysis and Phylogenetic Hierarchy
2.2. Evolutionary Length among DENV-3 Strains
3. Results
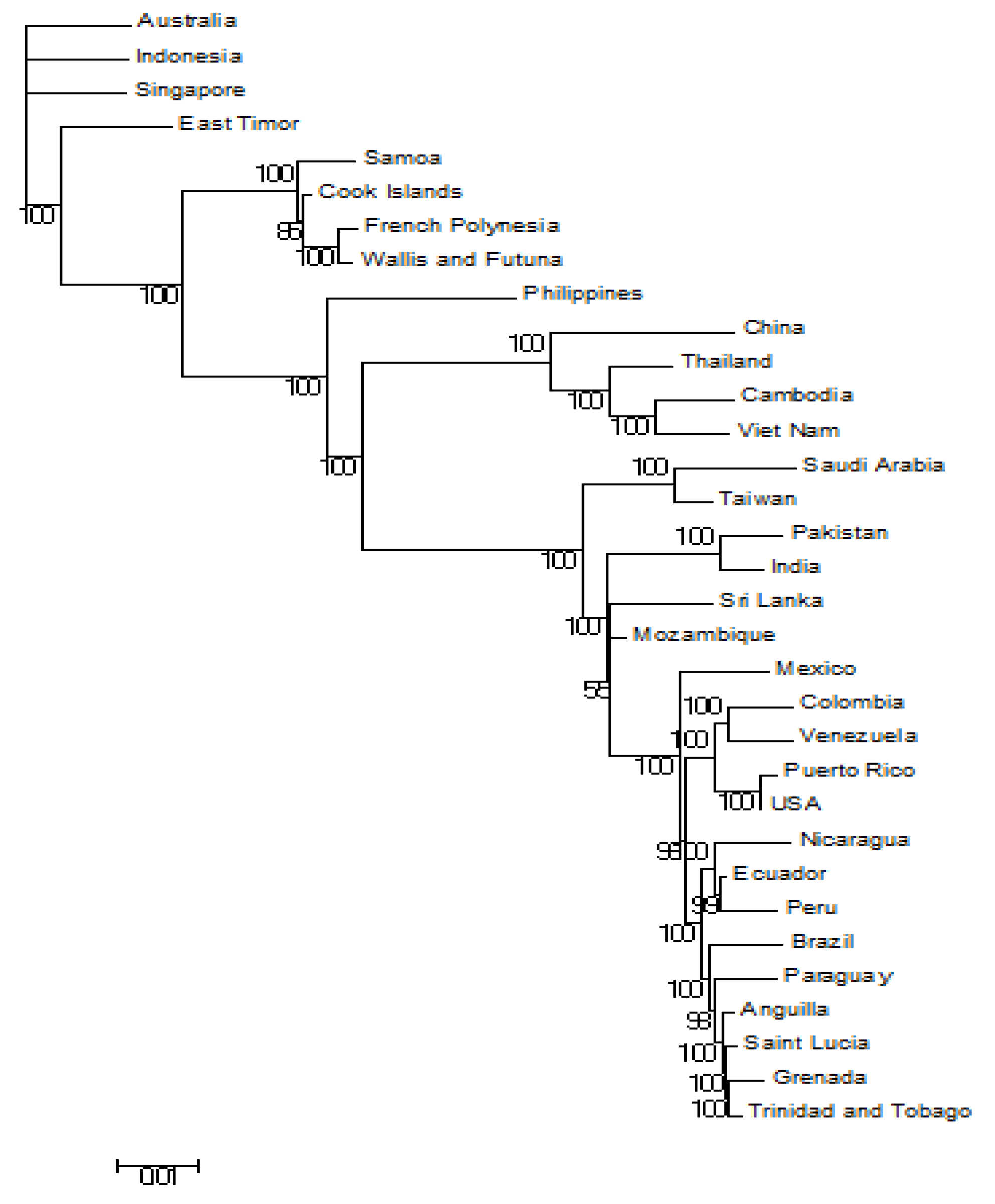
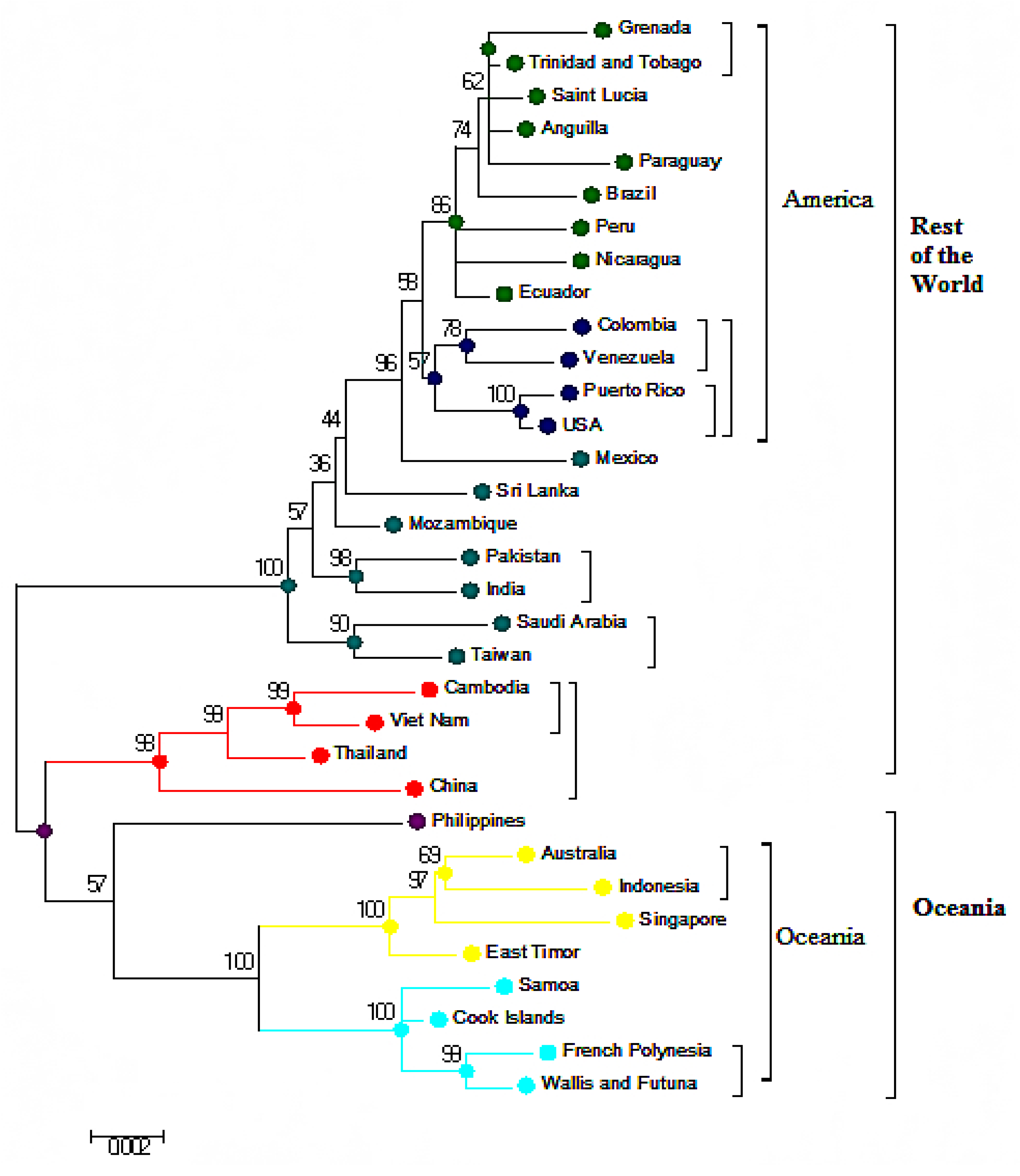
3.1. Phylogeneses and Phylogeographic Presentation
3.2. Bayesian Evolutionary Analysis (BEAST) of Whole-Genome DENV-3
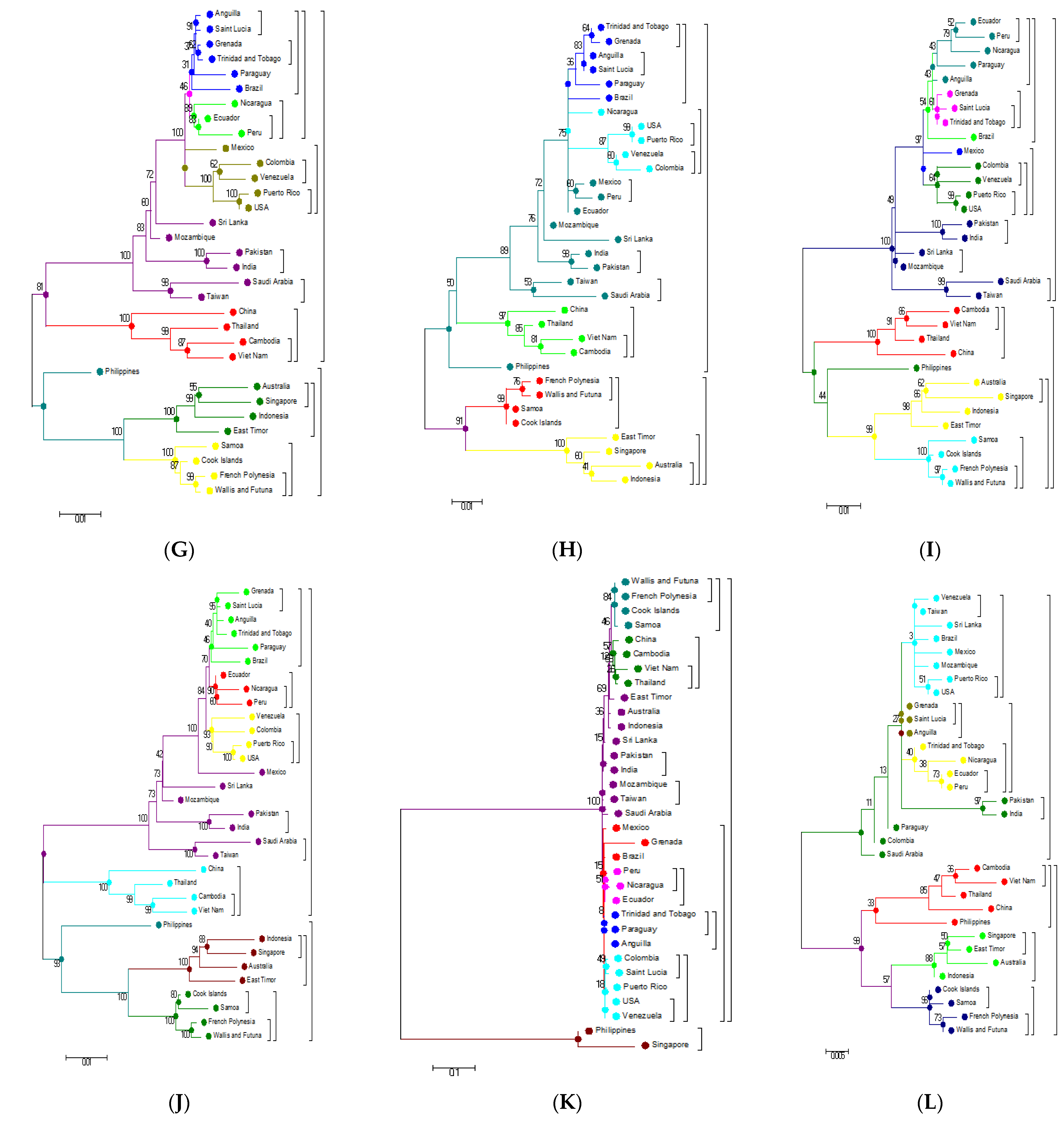
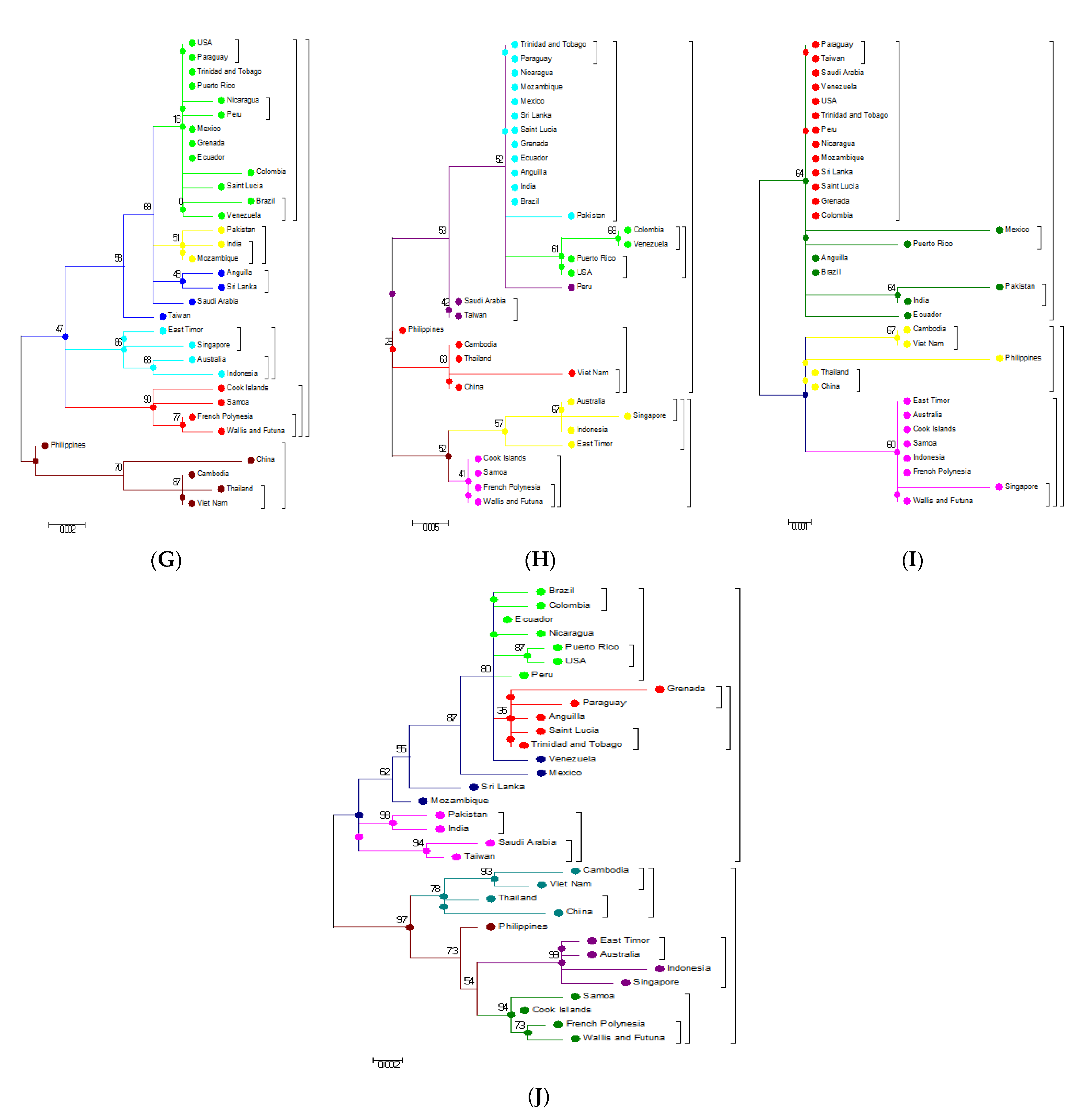
3.3. Phylogenetic Analysis Based on Amino Acids
3.4. Phylogenetic Analysis of Capsid (C) and Envelope (E) protein
3.5. Phylogenetic Analysis Based on Individual Proteins
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lindenbach, B.D.; Rice, C.M. Genetic interaction of flavivirus nonstructural proteins NS1 and NS4A as a determinant of replicase function. J. Virol. 1999, 736, 4611–4621. [Google Scholar] [CrossRef] [Green Version]
- Seligman, S.J. Constancy and diversity in the flavivirus fusion peptide. Virol. J. 2008, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilakis, N. Sylvatic Dengue: Evolution, Emergence, and Impact on Human Health. Ph.D. Thesis, 2007. Available online: https://utmb-ir.tdl.org/handle/2152.3/299 (accessed on 3 January 2020).
- Wahala, W.M.P.B.; Donaldson, E.F.; de Alwis, R.; Accavitti-Loper, M.A.; Baric, R.S.; de Silva, A.M. Natural strain variation and antibody neutralization of dengue serotype 3 viruses. PLoS Pathog. 2010, 6, e1000821. [Google Scholar] [CrossRef]
- Diallo, M.; Fernandez, Z.; Coffey, L.L.; Ba, Y.; Weaver, S.C.; Ortiz, D.; Mathiot, C.; Tesh, R.B.; Moncayo, A.C.; Sall, A.A. Potential role of sylvatic and domestic African mosquito species in dengue emergence. Am. J. Trop. Med. Hyg. 2005, 73, 445–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, J.S.; Gubler, D.J.; Petersen, L.R. Emerging flaviviruses: The spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat. Med. 2004, 10, S98–S109. [Google Scholar] [CrossRef]
- Gubler, D.J.; Clark, G.G. Dengue/dengue hemorrhagic fever: The emergence of a global health problem. Emer. Infect. Dis. 1995, 1, 55–57. [Google Scholar] [CrossRef]
- Kimura, R. Studies on dengue fever (VI). On the inoculation of dengue virus into mice. Nippon Igaku. 1944, 3379, 629–633. [Google Scholar]
- World Health Organization. Dengue Haemorrhagic Fever: Diagnosis, Treatment, Prevention and Control; World Health Organization: Geneva, Switzerland, 1997. [Google Scholar]
- Gubler, D.J. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 1998, 11, 480–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, S.C.; Vasilakis, N. Molecular evolution of dengue viruses: Contributions of phylogenetics to understanding the history and epidemiology of the preeminent arboviral disease. Infect. Genet. Evol. 2009, 9, 523–540. [Google Scholar] [CrossRef] [Green Version]
- Lanciotti, R.S.; Lewis, J.G.; Gubler, D.J.; Trent, D.W. Molecular evolution and epidemiology of dengue-3 viruses. J. General. Virol. 1994, 75, 65–75. [Google Scholar] [CrossRef]
- Aquino, V.H.; Amarilla, A.A.; Alfonso, H.L.; Batista, W.C.; Figureido, L.T.M. New genotype of dengue type 3 virus circulating in Brazil and Colombia showed a close relationship to old Asian viruses. PLoS ONE 2009, 4, e7299. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, A.R.; Milinkovitch, M.C. The metapopulation genetic algorithm: An efficient solution for the problem of large phylogeny estimation. Proc. Natl. Acad. Sci. USA 2002, 99, 10516–10521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.A.; Chang, G.J.; Lanciotti, R.S.; Kinney, R.M.; Mayer, L.W.; Trent, D.W. Phylogenetic relationships of dengue-2 viruses. Virology 1993, 197, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Klungthong, C.; Punak, R.; Mammen, M.P.; Zhang, C. Molecular genotyping of dengue viruses by phylogenetic analysis of the sequences of individual genes. J. Virol. Methods 2008, 154, 175–181. [Google Scholar] [CrossRef]
- Holmes, E.C.; Tio, P.H.; Perera, D.; Muhi, J.; Cardosa, J. Importation and co-circulation of multiple serotypes of dengue virus in Sarawak, Malaysia. Virus Res. 2009, 143, 1–5. [Google Scholar] [CrossRef]
- Lee, K.-S.; Lo, S.; Tan, S.S.-Y.; Chua, R.; Tan, L.-K.; Xu, H.; Ng, L.-C. Dengue virus surveillance in Singapore reveals high viral diversity through multiple introductions and in situ evolution. Infect. Genet. Evol. 2012, 12, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, L.B.; Cecílio, A.B.; Ferreira, G.P.; Drumond, B.P.; De Oliveira, J.G.; Bonjardim, C.A.; Ferreira, P.C.P.; Kroon, E.G. Dengue virus 3 genotype 1 associated with dengue fever and dengue hemorrhagic fever, Brazil. Emerg. Infect. Dis. 2008, 14, 314–316. [Google Scholar] [CrossRef]
- Miagostovich, M.P.; Dos Santos, F.B.; Fumian, T.M.; Guimarães, F.R.; Da Costa, E.V.; Tavares, F.N.; Coelho, J.O.; Nogueira, R.M.R. Complete genetic characterization of a Brazilian dengue virus type 3 strain isolated from a fatal outcome. Memórias do Instituto Oswaldo Cruz 2006, 101, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Suleman, M.; Faryal, R.; Alam, M.M.; Khurshid, A.; Sharif, S.; Shaukat, S.; Angez, M.; Umair, M.; Sufian, M.M.; Arshad, Y.; et al. Outbreak of dengue virus type-3 in Malakand, Pakistan 2015; A laboratory perspective. Acta Tropica. 2017, 169, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Angel, A.; Angel, B.; Joshi, A.P.; Baharia, R.K.; Rathore, S.; Joshi, V. First study of complete genome of Dengue-3 virus from Rajasthan, India: Genomic characterization, amino acid variations and phylogenetic analysis. Virol. Rep. 2016, 6, 32–40. [Google Scholar] [CrossRef] [Green Version]
- King, C.-C.; Chao, D.-Y.; Chien, L.-J.; Chang, G.-J.J.; Lin, T.-H.; Wu, Y.-C.; Huang, J.-H. Comparative analysis of full genomic sequences among different genotypes of dengue virus type 3. Virol. J. 2008, 5, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Dash, P.K.; Agarwal, S.; Shukla, J.; Parida, M.M.; Rao, P.V.L. Comparative complete genome analysis of dengue virus type 3 circulating in India between 2003 and 2008. J. Gen. Virol. 2011, 92, 1595–1600. [Google Scholar] [CrossRef]
- Pryor, M.J.; Carr, J.M.; Hocking, H.; Davidson, A.D.; Li, P.; Wright, P.J. Replication of dengue virus type 2 in human monocyte-derived macrophages: Comparisons of isolates and recombinant viruses with substitutions at amino acid 390 in the envelope glycoprotein. Am. J. Trop. Med. Hyg. 2001, 65, 427–434. [Google Scholar] [CrossRef]
- Sessions, O.M.; Andreas, W.; Kamaraj, U.S.; Choy, M.M.; Chow, A.; Chong, Y.; Ong, X.M.; Nagarajan, N.; Cook, A.R.; Ooi, E.E. Analysis of dengue virus genetic diversity during human and mosquito infection reveals genetic constraints. PLoS Negl. Trop. Dis. 2015, 9, e0004044. [Google Scholar] [CrossRef]
- Dorji, T.; Yoon, I.-K.; Holmes, E.C.; Wangchuk, S.; Tobgay, T.; Nisalak, A.; Chinnawirotpisan, P.; Sangkachantaranon, K.; Gibbons, R.V.; Jarman, R.G. Diversity and origin of dengue virus serotypes 1, 2, and 3, Bhutan. Emerg Infect Dis. 2009, 15, 1630–1632. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country | Name of Isolate | Genome (bp) | Year | Nucleotide Accession | Protein Accession | Reference |
|---|---|---|---|---|---|---|
| Anguilla | AI/BID-V2976/2001 | 10,663 | 2001 | FJ898462 | ACQ44501 | Direct Submission |
| Australia | Cairns 2008 | 10,707 | 2008 | JN406515 | AFN80339 | Direct Submission |
| Cambodia | KH/BID-V2053/2008 | 10,648 | 2008 | FJ639715 | ACL99233 | Direct Submission |
| Colombia | CO/BID-V3405/2007 | 10,659 | 2007 | GQ868578 | ACW83006 | Direct Submission |
| East Timor | Hu/TL018NIID/2005 | 10,707 | 2005 | AB214879 | BAE48725 | Direct Submission |
| Ecuador | EC/BID-V2975/2000 | 10,663 | 2000 | FJ898457 | ACQ44496 | Direct Submission |
| French Polynesia | PF96/150296-46183 | 10,671 | 1994 | JQ920479 | AFY10043 | Direct Submission |
| India | DEL-72 | 10,680 | 2008 | GQ466079 | ADM63678 | Direct Submission |
| Mexico | MX/BID-V2989/2007 | 10,663 | 2007 | FJ898442 | ACQ44481 | Direct Submission |
| Viet Nam | VN/BID-V1911/2008 | 10,637 | 2008 | FJ547066 | ACL98983 | Direct Submission |
| Mozambique | MZ/BID-V2418/1985 | 10,663 | 1985 | FJ882575 | ACQ44384 | Direct Submission |
| Paraguay | PAR 5532-07 | 10,707 | 2007 | HQ235027 | AEF65939 | Direct Submission |
| Saint Lucia | LC/BID-V2979/2001 | 10,660 | 2001 | FJ898463 | ACQ44502 | Direct Submission |
| Singapore | SGEHI(D3)0040Y09 | 10,250 | 2009/01 | GU370052 | ADC92353 | Direct Submission |
| Sri Lanka | LK/BID-V2409/1997 | 10,682 | 1997 | GQ252674 | ACS32036 | Direct Submission |
| Taiwan | 99TW628 | 10,707 | 1999 | DQ675533 | ABG73599 | Direct Submission |
| Thailand | TH/BID-V2318/2001 | 10,629 | 2001 | FJ687448 | ACN42695 | Direct Submission |
| Trinidad and Tobago | TT/BID-V2982/2002 | 10,663 | 2002 | FJ898459 | ACQ44498 | Direct Submission |
| USA | US/BID-V1620/2005 | 10,648 | 2005 | FJ182010 | ACH99657 | Direct Submission |
| Venezuela | VE/BID-V2267/2008, | 10,654 | 2008 | FJ639826 | ACL99113 | Direct Submission |
| Samoa | WS/BID-V2973/1995 | 10,663 | 1995 | FJ898456 | ACQ44495 | Direct Submission |
| Wallis and Futuna | WF95/090595-2448 | 10,671 | 9/5/1995 | JQ920489 | AFY10053 | Direct Submission |
| Cook Islands | CK/BID-V2972/1991 | 10,663 | 1991 | FJ898455 | ACQ44494 | Direct Submission |
| Brazil | BR/AL95/2009 | 10,707 | 2009 | JF808120 | AFK83755 | Direct Submission |
| China | YN01 | 10,707 | 2013 | KF824902 | AHI17474 | Direct Submission |
| Grenada | GD/BID-V3930/2002 | 10,653 | 2002 | KF955505 | AHG23270 | Direct Submission |
| Indonesia | MKS-WS79b | 10,707 | 29/03/2010 | KC762693 | AHG06377 | Direct Submission |
| Nicaragua | NI/BID-V7658/2012 | 10,569 | 4/7/1905 | KF973480 | AHC98451 | Direct Submission |
| Pakistan | Pakistan/56/2008 | 10,675 | 2008 | KF041254 | AHC72426 | Direct Submission |
| Peru | PE/BID-V7289/2008 | 10,693 | 2008 | KJ189301 | AHI43684 | Direct Submission |
| Philippines | VIROAF7 | 10,267 | 1964 | KM190937 | AIG60036 | Direct Submission |
| Puerto Rico | PR/BID-V1728/2006 | 10,645 | 2006 | KF955456 | AHG23221 | Direct Submission |
| Saudi Arabia | Jeddah-2014 | 10,635 | 26/01/2014 | KJ830751 | AIH13925 | Direct Submission |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amir, M.; Hussain, A.; Asif, M.; Ahmed, S.; Alam, H.; Moga, M.A.; Cocuz, M.E.; Marceanu, L.; Blidaru, A. Full-Length Genome and Partial Viral Genes Phylogenetic and Geographical Analysis of Dengue Serotype 3 Isolates. Microorganisms 2021, 9, 323. https://doi.org/10.3390/microorganisms9020323
Amir M, Hussain A, Asif M, Ahmed S, Alam H, Moga MA, Cocuz ME, Marceanu L, Blidaru A. Full-Length Genome and Partial Viral Genes Phylogenetic and Geographical Analysis of Dengue Serotype 3 Isolates. Microorganisms. 2021; 9(2):323. https://doi.org/10.3390/microorganisms9020323
Chicago/Turabian StyleAmir, Muhammad, Abrar Hussain, Muhammad Asif, Sagheer Ahmed, Hina Alam, Marius Alexandru Moga, Maria Elena Cocuz, Luigi Marceanu, and Alexandru Blidaru. 2021. "Full-Length Genome and Partial Viral Genes Phylogenetic and Geographical Analysis of Dengue Serotype 3 Isolates" Microorganisms 9, no. 2: 323. https://doi.org/10.3390/microorganisms9020323