
A Genomic and Transcriptomic Study on the DDT-Resistant Trichoderma hamatum FBL 587: First Genetic Data into Mycoremediation Strategies for DDT-Polluted Sites

 , ,
, ,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
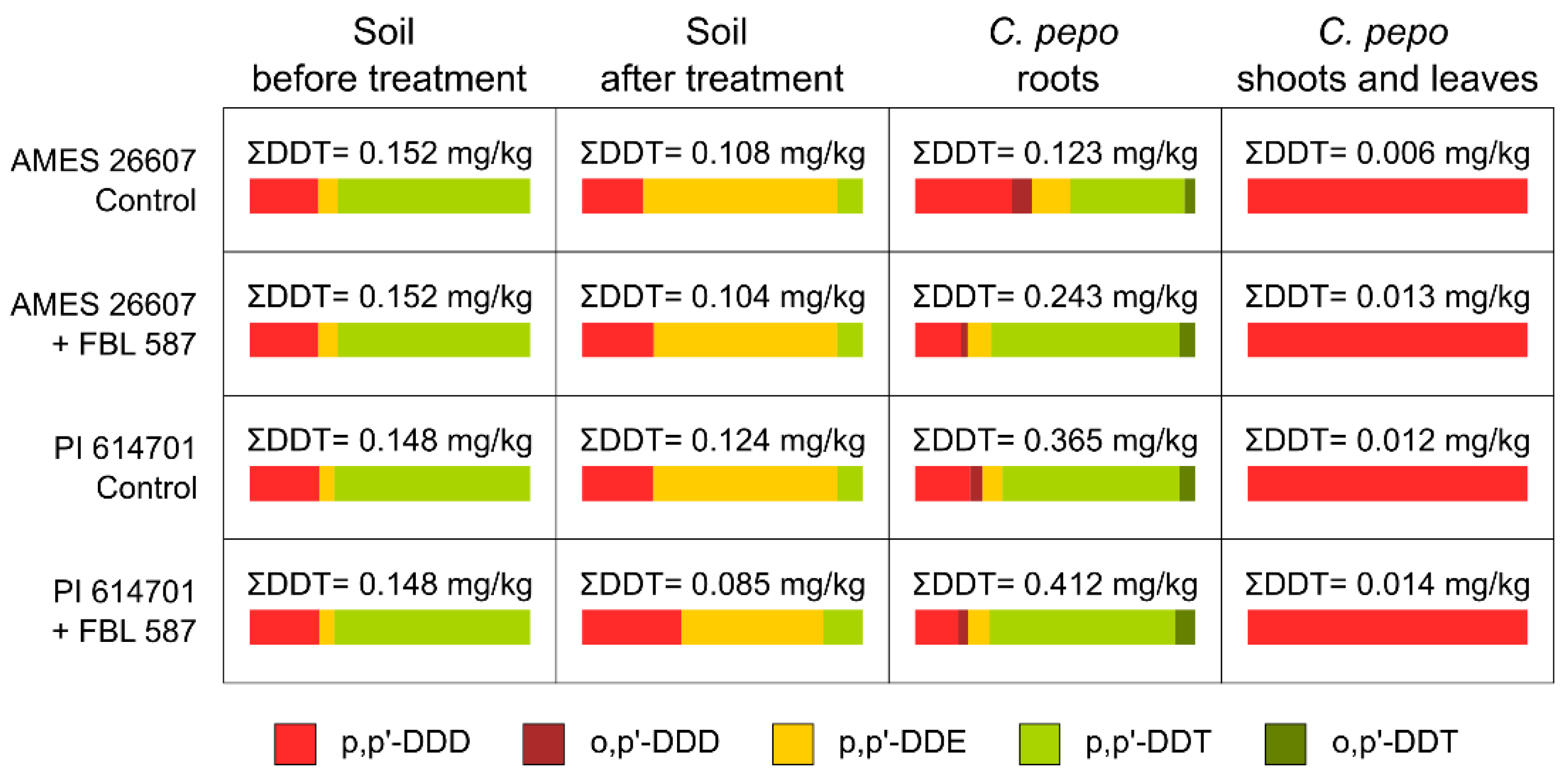
2.1. DDT Degradation Process in Soil by T. hamatum FBL 587
2.2. Genomic DNA Extraction, Whole Genome Sequencing and Bioinformatics Analyses
2.3. RNA Extraction and mRNA Sequencing
2.4. Transcriptome Profiling with RNA-Seq Approach
2.5. Accession Numbers of the Genome and Transcriptome Sequences
3. Results
3.1. DDT Degradation Process in Soil by T. hamatum FBL 587
3.2. Genome Properties of T. hamatum FBL 587
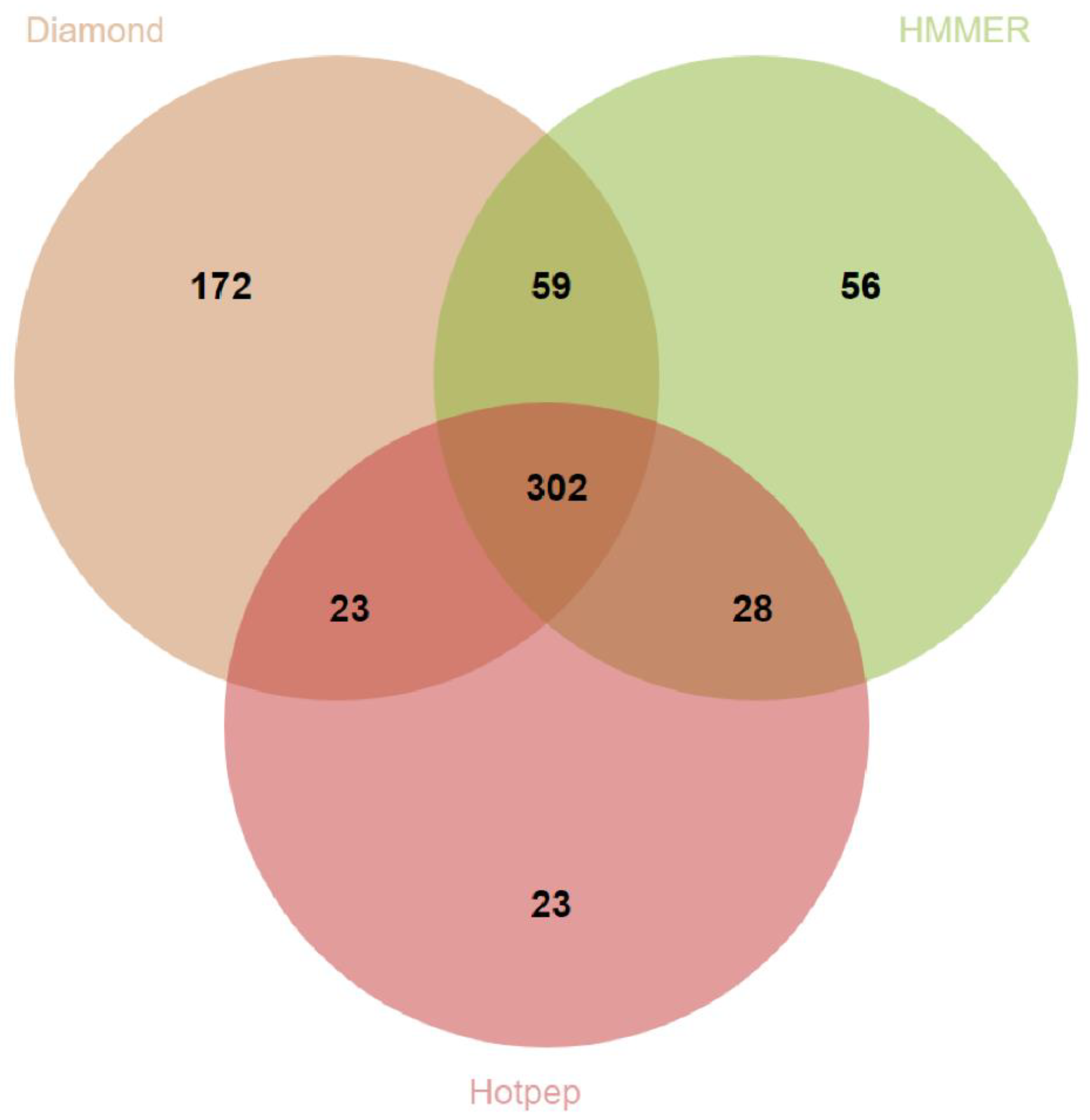
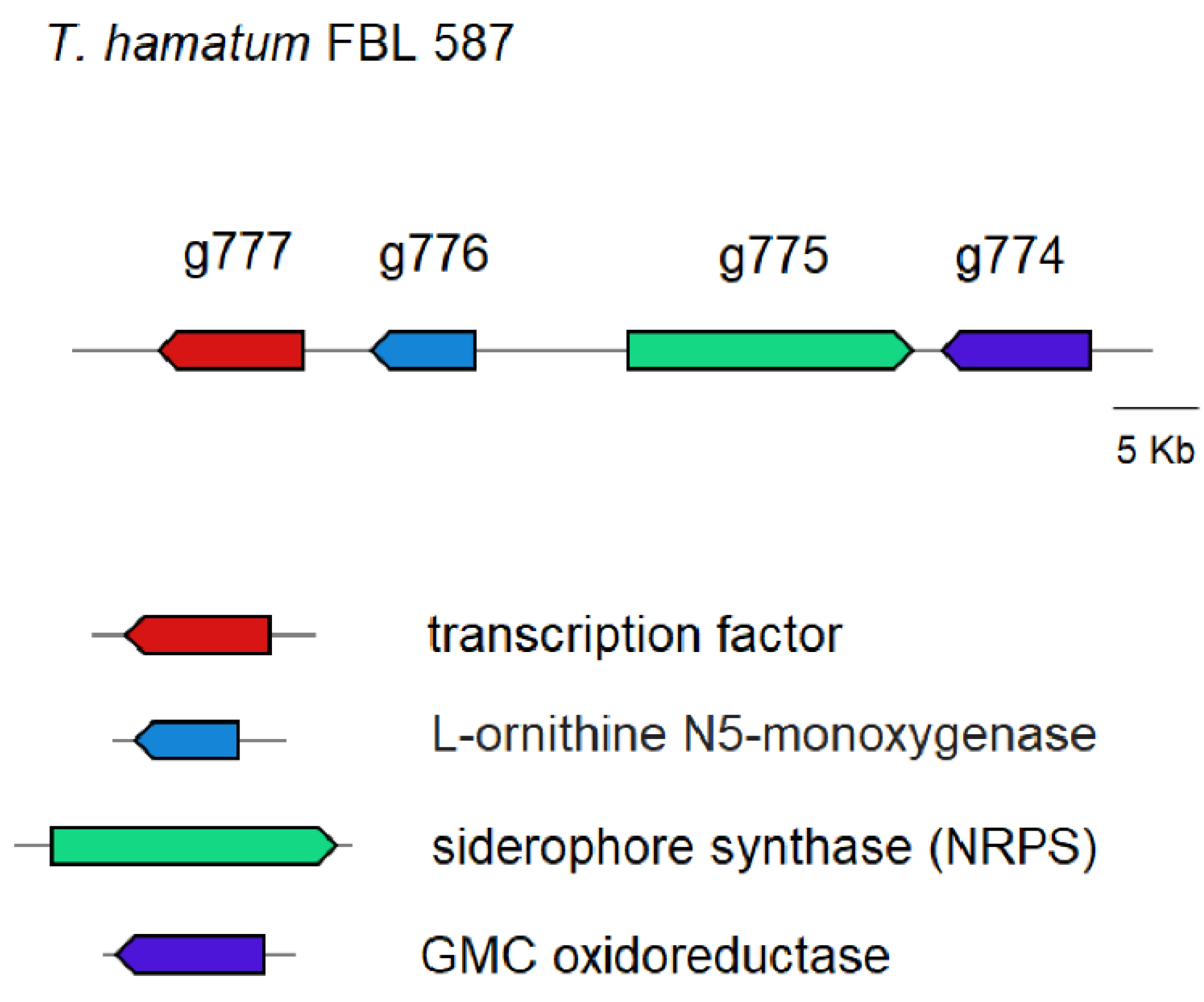
3.3. Prediction of Genes Encoding for CAZymes, SMs and Siderophores
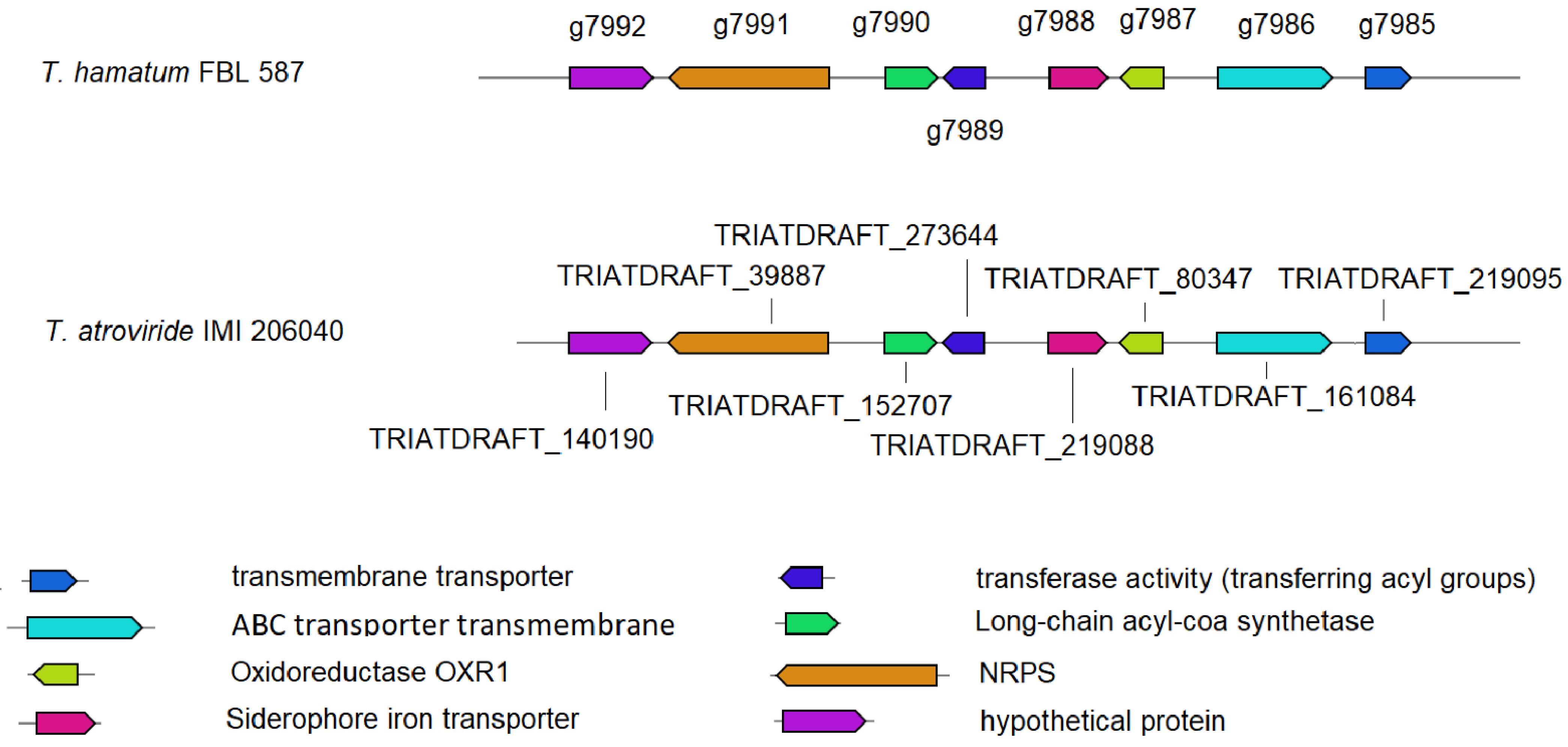
3.4. Prediction of Genes Involved in the Metabolism of DDT
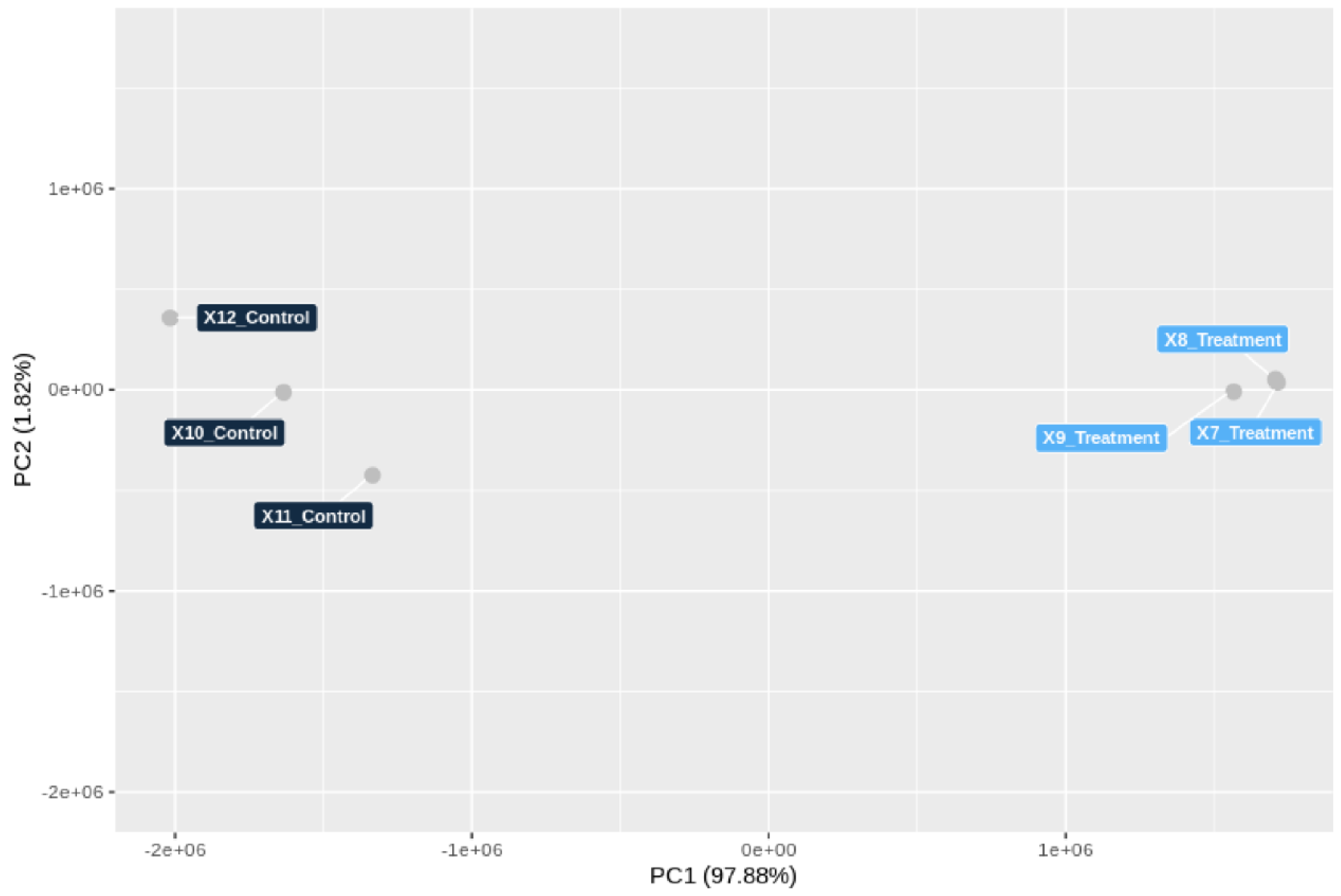
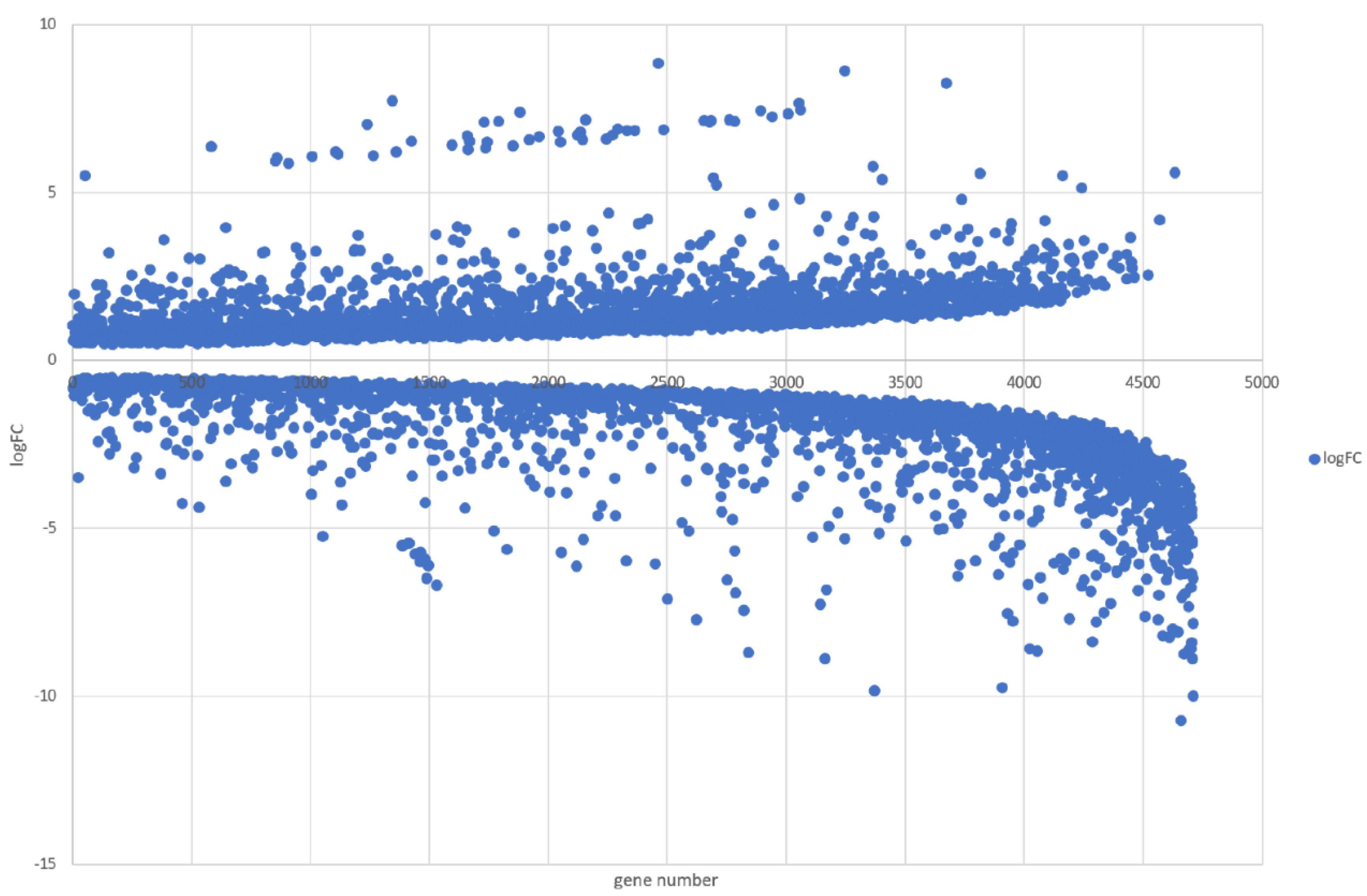
3.5. Genome-Wide Transcriptomic Responses of T. hamatum FBL 587 during DDT Exposure
3.6. Transcriptomic Analysis of DDT Tolerance Genes by T. hamatum FBL 587
3.7. Transcriptomic Analysis of DDT Transforming Genes by T. hamatum FBL 587
4. Discussion
4.1. Genome Properties of T. hamatum FBL 587
4.2. Transcriptomic Analysis of DDT Tolerance Genes by T. hamatum FBL 587
4.3. Transcriptomic Analysis of DDT Transforming Genes by T. hamatum FBL 587
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hung, H.; Katsoyiannis, A.A.; Guardans, R. Ten Years of Global Monitoring under the Stockholm Convention on Persistent Organic Pollutants (POPs): Trends, Sources and Transport Modelling. Environ. Pollut. 2016, 217, 1–3. [Google Scholar] [CrossRef]
- Silva, V.; Mol, H.G.J.; Zomer, P.; Tienstra, M.; Ritsema, C.J.; Geissen, V. Pesticide Residues in European Agricultural Soils—A Hidden Reality Unfolded. Sci. Total Environ. 2019, 653, 1532–1545. [Google Scholar] [CrossRef]
- Malusá, E.; Tartanus, M.; Danelski, W.; Miszczak, A.; Szustakowska, E.; Kicińska, J.; Furmanczyk, E.M. Monitoring of DDT in Agricultural Soils under Organic Farming in Poland and the Risk of Crop Contamination. Environ. Manag. 2020, 66, 916–929. [Google Scholar] [CrossRef]
- Venice, F.; Davolos, D.; Spina, F.; Poli, A.; Prigione, V.P.; Varese, G.C.; Ghignone, S. Genome Sequence of Trichoderma Lixii MUT3171, a Promising Strain for Mycoremediation of PAH-Contaminated Sites. Microorganisms 2020, 8, 1258. [Google Scholar] [CrossRef]
- Russo, F.; Ceci, A.; Pinzari, F.; Siciliano, A.; Guida, M.; Malusà, E.; Tartanus, M.; Miszczak, A.; Maggi, O.; Persiani, A.M. Bioremediation of Dichlorodiphenyltrichloroethane (DDT)-Contaminated Agricultural Soils: Potential of Two Autochthonous Saprotrophic Fungal Strains. Appl. Environ. Microbiol. 2019, 85, e01720-19. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Singh, P.C.; Mishra, A.; Chauhan, P.S.; Dwivedi, S.; Bais, R.T.; Tripathi, R.D. Trichoderma: A Potential Bioremediator for Environmental Clean Up. Clean Technol. Environ. Policy 2013, 15, 541–550. [Google Scholar] [CrossRef]
- Zafra, G.; Moreno-Montaño, A.; Absalón, Á.E.; Cortés-Espinosa, D.V. Degradation of Polycyclic Aromatic Hydrocarbons in Soil by a Tolerant Strain of Trichoderma Asperellum. Environ. Sci. Pollut. Res. 2015, 22, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Zafra, G.; Cortés-Espinosa, D.V. Biodegradation of Polycyclic Aromatic Hydrocarbons by Trichoderma Species: A Mini Review. Environ. Sci. Pollut. Res. 2015, 22, 19426–19433. [Google Scholar] [CrossRef] [PubMed]
- Sang, H.; Hulvey, J.P.; Green, R.; Xu, H.; Im, J.; Chang, T.; Jung, G. A Xenobiotic Detoxification Pathway through Transcriptional Regulation in Filamentous Fungi. mBio 2018, 9, e00457-18. [Google Scholar] [CrossRef] [Green Version]
- Jeong, C.-B.; Kim, H.-S.; Kang, H.-M.; Lee, J.-S. ATP-Binding Cassette (ABC) Proteins in Aquatic Invertebrates: Evolutionary Significance and Application in Marine Ecotoxicology. Aquat. Toxicol. 2017, 185, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Olicón-Hernández, D.R.; González-López, J.; Aranda, E. Overview on the Biochemical Potential of Filamentous Fungi to Degrade Pharmaceutical Compounds. Front. Microbiol. 2017, 8, 1792. [Google Scholar] [CrossRef]
- Druzhinina, I.S.; Seidl-Seiboth, V.; Herrera-Estrella, A.; Horwitz, B.A.; Kenerley, C.M.; Monte, E.; Mukherjee, P.K.; Zeilinger, S.; Grigoriev, I.V.; Kubicek, C.P. Trichoderma: The Genomics of Opportunistic Success. Nat. Rev. Microbiol. 2011, 9, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Liu, B.; Su, Y.; Hu, Y.; Hong, Y.; Yi, X.; Chen, L.; Su, S.; Chu, J.S.C.; Chen, N.; et al. Genomics Insights into Different Cellobiose Hydrolysis Activities in Two Trichoderma Hamatum Strains. Microb. Cell Fact. 2017, 16, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studholme, D.J.; Harris, B.; Le Cocq, K.; Winsbury, R.; Perera, V.; Ryder, L.; Ward, J.L.; Beale, M.H.; Thornton, C.R.; Grant, M. Investigating the Beneficial Traits of Trichoderma Hamatum GD12 for Sustainable Agriculture—Insights from Genomics. Front. Plant Sci. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Kubicek, C.P.; Steindorff, A.S.; Chenthamara, K.; Manganiello, G.; Henrissat, B.; Zhang, J.; Cai, F.; Kopchinskiy, A.G.; Kubicek, E.M.; Kuo, A.; et al. Evolution and Comparative Genomics of the Most Common Trichoderma Species. BMC Genom. 2019, 20, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.C.; Zeeb, B.A. Plant Phylogeny and the Remediation of Persistent Organic Pollutants. In Phytoremediation; Willey, N., Ed.; Methods in Biotechnology; Humana Press: Totowa, NJ, USA, 2007; Volume 23, pp. 71–87. ISBN 978-1-58829-541-5. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.Bioinformatics.Babraham.Ac.Uk/Projects (accessed on 24 January 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Nikolenko, S.I.; Korobeynikov, A.I.; Alekseyev, M.A. BayesHammer: Bayesian Clustering for Error Correction in Single-Cell Sequencing. BMC Genom. 2013, 14, S7. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and Rapid Annotation of Ribosomal RNA Genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. TRNAscan-SE On-Line: Integrating Search and Context for Analysis of Transfer RNA Genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2018. Available online: http://Repeatmasker.Org/ (accessed on 1 July 2020).
- Manchanda, N.; Portwood, J.L.; Woodhouse, M.R.; Seetharam, A.S.; Lawrence-Dill, C.J.; Andorf, C.M.; Hufford, M.B. GenomeQC: A Quality Assessment Tool for Genome Assemblies and Gene Structure Annotations. BMC Genom. 2020, 21, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoff, K.J.; Stanke, M. Predicting Genes in Single Genomes with AUGUSTUS. Curr. Protoc. Bioinform. 2019, 65, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A Rapid Functional Annotation Web Server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. DbCAN2: A Meta Server for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving Cluster Detection and Comparison Capabilities. Nucleic Acids Res. 2021, gkab335. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kautsar, S.A.; Medema, M.H.; Weber, T. The AntiSMASH Database Version 3: Increased Taxonomic Coverage and New Query Features for Modular Enzymes. Nucleic Acids Res. 2021, 49, D639–D643. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Rau, A.; Gallopin, M.; Celeux, G.; Jaffrézic, F. Data-Based Filtering for Replicated High-Throughput Transcriptome Sequencing Experiments. Bioinformatics 2013, 29, 2146–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroncelli, R.; Zapparata, A.; Piaggeschi, G.; Sarrocco, S.; Vannacci, G. Draft Whole-Genome Sequence of Trichoderma Gamsii T6085, a Promising Biocontrol Agent of Fusarium Head Blight on Wheat. Genome Announc. 2016, 4, e01747-15. [Google Scholar] [CrossRef] [Green Version]
- Shi-Kunne, X.; Seidl, M.F.; Faino, L.; Thomma, B.P.H.J. Draft Genome Sequence of a Strain of Cosmopolitan Fungus Trichoderma Atroviride. Genome Announc. 2015, 3, e00287-15. [Google Scholar] [CrossRef] [Green Version]
- Chooi, Y.-H.; Muria-Gonzalez, M.J.; Mead, O.L.; Solomon, P.S. SnPKS19 Encodes the Polyketide Synthase for Alternariol Mycotoxin Biosynthesis in the Wheat Pathogen Parastagonospora Nodorum. Appl. Environ. Microbiol. 2015, 81, 5309–5317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonsch, B.; Belt, V.; Bartel, C.; Duensing, N.; Koziol, M.; Lazarus, C.M.; Bailey, A.M.; Simpson, T.J.; Cox, R.J. Identification of Genes Encoding Squalestatin S1 Biosynthesis and in Vitro Production of New Squalestatin Analogues. Chem. Commun. 2016, 52, 6777–6780. [Google Scholar] [CrossRef] [Green Version]
- Godio, R.P.; Fouces, R.; Martín, J.F. A Squalene Epoxidase Is Involved in Biosynthesis of Both the Antitumor Compound Clavaric Acid and Sterols in the Basidiomycete H. Sublateritium. Chem. Biol. 2007, 14, 1334–1346. [Google Scholar] [CrossRef] [Green Version]
- Godio, R.P.; Martín, J.F. Modified Oxidosqualene Cyclases in the Formation of Bioactive Secondary Metabolites: Biosynthesis of the Antitumor Clavaric Acid. Fungal Genet. Biol. 2009, 46, 232–242. [Google Scholar] [CrossRef]
- Chen, L.-H.; Lin, C.-H.; Chung, K.-R. A Nonribosomal Peptide Synthetase Mediates Siderophore Production and Virulence in the Citrus Fungal Pathogen Alternaria Alternata: Nonribosomal Peptide Synthetase of Alternaria. Mol. Plant Pathol. 2013, 14, 497–505. [Google Scholar] [CrossRef]
- Chen, L.-H.; Yang, S.L.; Chung, K.-R. Resistance to Oxidative Stress via Regulating Siderophore-Mediated Iron Acquisition by the Citrus Fungal Pathogen Alternaria Alternata. Microbiology 2014, 160, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Horwitz, B.A.; Herrera-Estrella, A.; Schmoll, M.; Kenerley, C.M. Trichoderma Research in the Genome Era. Annu. Rev. Phytopathol. 2013, 51, 105–129. [Google Scholar] [CrossRef] [PubMed]
- Chadha, S.; Mehetre, S.T.; Bansal, R.; Kuo, A.; Aerts, A.; Grigoriev, I.V.; Druzhinina, I.S.; Mukherjee, P.K. Genome-Wide Analysis of Cytochrome P450s of Trichoderma Spp.: Annotation and Evolutionary Relationships. Fungal Biol. Biotechnol. 2018, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, G.S.; Adriani, P.P.; Ribeiro, J.A.; Morisseau, C.; Hammock, B.D.; Dias, M.V.B.; Chambergo, F.S. The Molecular Structure of an Epoxide Hydrolase from Trichoderma Reesei in Complex with Urea or Amide-Based Inhibitors. Int. J. Biol. Macromol. 2019, 129, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Vicente, I.; Baroncelli, R.; Morán-Diez, M.E.; Bernardi, R.; Puntoni, G.; Hermosa, R.; Monte, E.; Vannacci, G.; Sarrocco, S. Combined Comparative Genomics and Gene Expression Analyses Provide Insights into the Terpene Synthases Inventory in Trichoderma. Microorganisms 2020, 8, 1603. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ghirardo, A.; Weber, B.; Schnitzler, J.-P.; Benz, J.P.; Rosenkranz, M. Trichoderma Species Differ in Their Volatile Profiles and in Antagonism toward Ectomycorrhiza Laccaria Bicolor. Front. Microbiol. 2019, 10, 891. [Google Scholar] [CrossRef] [PubMed]
- Bian, G.; Han, Y.; Hou, A.; Yuan, Y.; Liu, X.; Deng, Z.; Liu, T. Releasing the Potential Power of Terpene Synthases by a Robust Precursor Supply Platform. Metab. Eng. 2017, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, G.-Y.; Li, X.-Z.; Hu, M.; Wang, B.-Y.; Ruan, B.-H.; Zhou, H.; Zhao, L.-X.; Zhou, J.; Ding, Z.-T.; et al. Phytotoxic, Antibacterial, and Antioxidant Activities of Mycotoxins and Other Metabolites from Trichoderma sp. Nat. Prod. Res. 2017, 31, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-F.; Li, G.-H.; Zhang, K.-Q. Non-Volatile Metabolites from Trichoderma spp. Metabolites 2019, 9, 58. [Google Scholar] [CrossRef] [Green Version]
- Fanelli, F.; Liuzzi, V.C.; Logrieco, A.F.; Altomare, C. Genomic Characterization of Trichoderma Atrobrunneum (T. Harzianum Species Complex) ITEM 908: Insight into the Genetic Endowment of a Multi-Target Biocontrol Strain. BMC Genom. 2018, 19, 1–18. [Google Scholar] [CrossRef]
- Eisendle, M.; Schrettl, M.; Kragl, C.; Müller, D.; Illmer, P.; Haas, H. The Intracellular Siderophore Ferricrocin Is Involved in Iron Storage, Oxidative-Stress Resistance, Germination, and Sexual Development in Aspergillus Nidulans. Eukaryot. Cell 2006, 5, 1596–1603. [Google Scholar] [CrossRef] [Green Version]
- Črešnar, B.; Petrič, Š. Cytochrome P450 Enzymes in the Fungal Kingdom. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.-H.; Xiong, A.-S.; Xue, Y.; Fu, X.-Y.; Gao, F.; Zhao, W.; Tian, Y.-S.; Yao, Q.-H. Microbial Biodegradation of Polyaromatic Hydrocarbons. FEMS Microbiol. Rev. 2008, 32, 927–955. [Google Scholar] [CrossRef] [Green Version]
- Lah, L.; Podobnik, B.; Novak, M.; Korošec, B.; Berne, S.; Vogelsang, M.; Kraševec, N.; Zupanec, N.; Stojan, J.; Bohlmann, J.; et al. The Versatility of the Fungal Cytochrome P450 Monooxygenase System Is Instrumental in Xenobiotic Detoxification: Fungal P450 Systems in Xenobiotic Detoxification. Mol. Microbiol. 2011, 81, 1374–1389. [Google Scholar] [CrossRef] [Green Version]
- Faber, B.W.; van Gorcom, R.F.M.; Duine, J.A. Purification and Characterization of Benzoate-Para-Hydroxylase, a Cytochrome P450 (CYP53A1), from Aspergillus Niger. Arch. Biochem. Biophys. 2001, 394, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.D. Fungal Degradation of Benzoic Acid and Related Compounds. World J. Microbiol. Biotechnol. 1993, 9, 9–16. [Google Scholar] [CrossRef]
- Villalobos-Escobedo, J.M.; Herrera-Estrella, A.; Carreras-Villaseñor, N. The Interaction of Fungi with the Environment Orchestrated by RNAi. Mycologia 2016, 108, 556–571. [Google Scholar] [CrossRef] [PubMed]
- Lumjuan, N.; Wicheer, J.; Leelapat, P.; Choochote, W.; Somboon, P. Identification and Characterisation of Aedes Aegypti Aldehyde Dehydrogenases Involved in Pyrethroid Metabolism. PLoS ONE 2014, 9, e102746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouanneau, Y.; Micoud, J.; Meyer, C. Purification and Characterization of a Three-Component Salicylate 1-Hydroxylase from Sphingomonas Sp. Strain CHY-1. Appl. Environ. Microbiol. 2007, 73, 7515–7521. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Estimated Value |
|---|---|
| Number reads | 6,329,382 |
| Number used reads | 6,251,876 |
| Number of contigs (≥500 bp) | 1354 |
| Largest contig | 347,610 |
| N50 reads | 69,288 |
| L50 contigs | 166 |
| Predicted genes | 10,944 |
| Genome size | 38,965,975 bp |
| GC content | 48.54% |
| tRNA | 230 |
| 8S rRNA | 50 |
| 18S rRNA | 1 |
| 28S rRNA | 1 |
| Repeat class/family | |
| Simple repeats | 399,867 (1%) |
| Low complexity | 79,381 (0.2%) |
| Completeness | 98.5% |
| Fragmented | 0.7% |
| Missing | 0.2% |
| Complete and duplicated copy | 0.6% |
| Genome | NRPS, T1PKS | NRPS | NRPS-Like | T1PKS | Terpene | Total |
|---|---|---|---|---|---|---|
| Trichoderma asperellum CBS 433.97 | 6 | 10 | 10 | 11 | 11 | 48 |
| Trichoderma atroviride IMI 206040 | 4 | 10 | 10 | 12 | 8 | 44 |
| Trichoderma gamsii T6085 | 4 | 9 | 11 | 12 | 7 | 43 |
| Trichoderma hamatum GD12 | 5 | 5 | 6 | 7 | 5 | 28 |
| Trichoderma hamatum FBL 587 | 4 | 4 | 5 | 8 | 6 | 27 |
| Trichoderma lixii MUT3171 | 9 | 4 | 7 | 19 | 8 | 47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davolos, D.; Russo, F.; Canfora, L.; Malusà, E.; Tartanus, M.; Furmanczyk, E.M.; Ceci, A.; Maggi, O.; Persiani, A.M. A Genomic and Transcriptomic Study on the DDT-Resistant Trichoderma hamatum FBL 587: First Genetic Data into Mycoremediation Strategies for DDT-Polluted Sites. Microorganisms 2021, 9, 1680. https://doi.org/10.3390/microorganisms9081680
Davolos D, Russo F, Canfora L, Malusà E, Tartanus M, Furmanczyk EM, Ceci A, Maggi O, Persiani AM. A Genomic and Transcriptomic Study on the DDT-Resistant Trichoderma hamatum FBL 587: First Genetic Data into Mycoremediation Strategies for DDT-Polluted Sites. Microorganisms. 2021; 9(8):1680. https://doi.org/10.3390/microorganisms9081680
Chicago/Turabian StyleDavolos, Domenico, Fabiana Russo, Loredana Canfora, Eligio Malusà, Małgorzata Tartanus, Ewa Maria Furmanczyk, Andrea Ceci, Oriana Maggi, and Anna Maria Persiani. 2021. "A Genomic and Transcriptomic Study on the DDT-Resistant Trichoderma hamatum FBL 587: First Genetic Data into Mycoremediation Strategies for DDT-Polluted Sites" Microorganisms 9, no. 8: 1680. https://doi.org/10.3390/microorganisms9081680
APA StyleDavolos, D., Russo, F., Canfora, L., Malusà, E., Tartanus, M., Furmanczyk, E. M., Ceci, A., Maggi, O., & Persiani, A. M. (2021). A Genomic and Transcriptomic Study on the DDT-Resistant Trichoderma hamatum FBL 587: First Genetic Data into Mycoremediation Strategies for DDT-Polluted Sites. Microorganisms, 9(8), 1680. https://doi.org/10.3390/microorganisms9081680