


Identification of Candidate Genes and eQTLs Related to Porcine Reproductive Function
 ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources
2.2. RNA-Seq Data Analyses
2.3. SNP Calling and Quality Control of RNA-Seq Samples
2.4. Covariates for eQTL Mapping
2.5. cis-eQTL Mapping and Fine-Mapping
2.6. WGCNA
2.7. Gene Functional Enrichment Analysis
3. Results
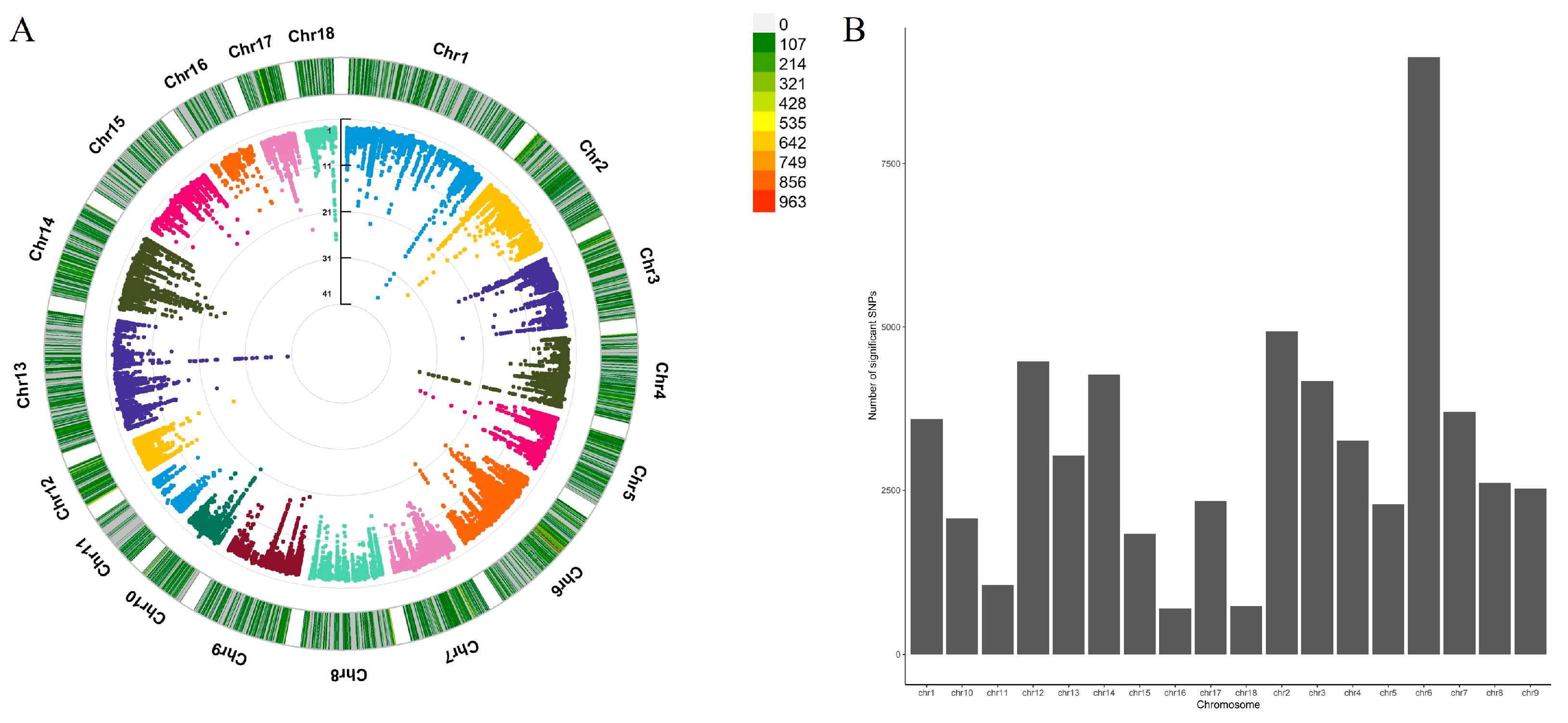
3.1. cis-eQTL Analysis
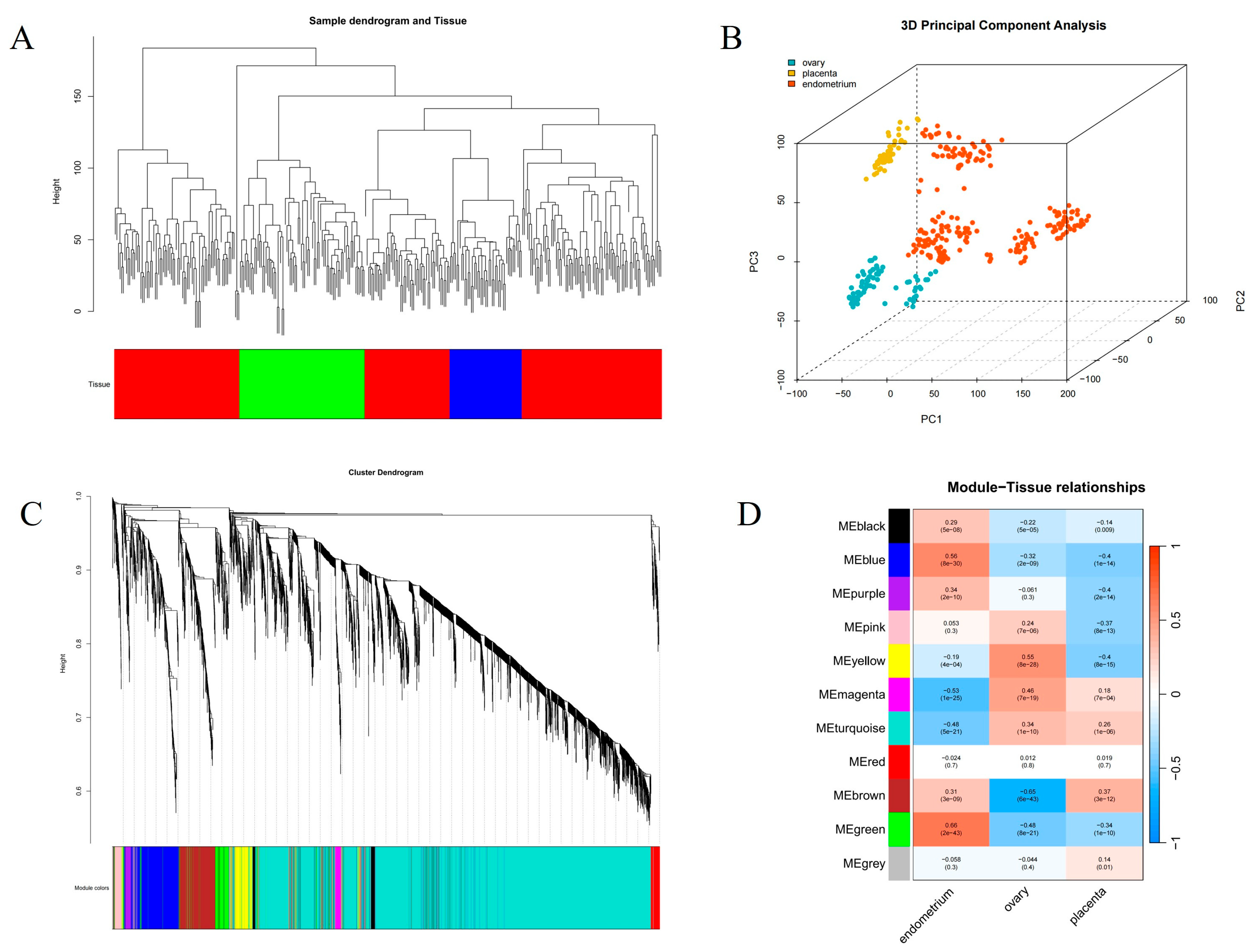
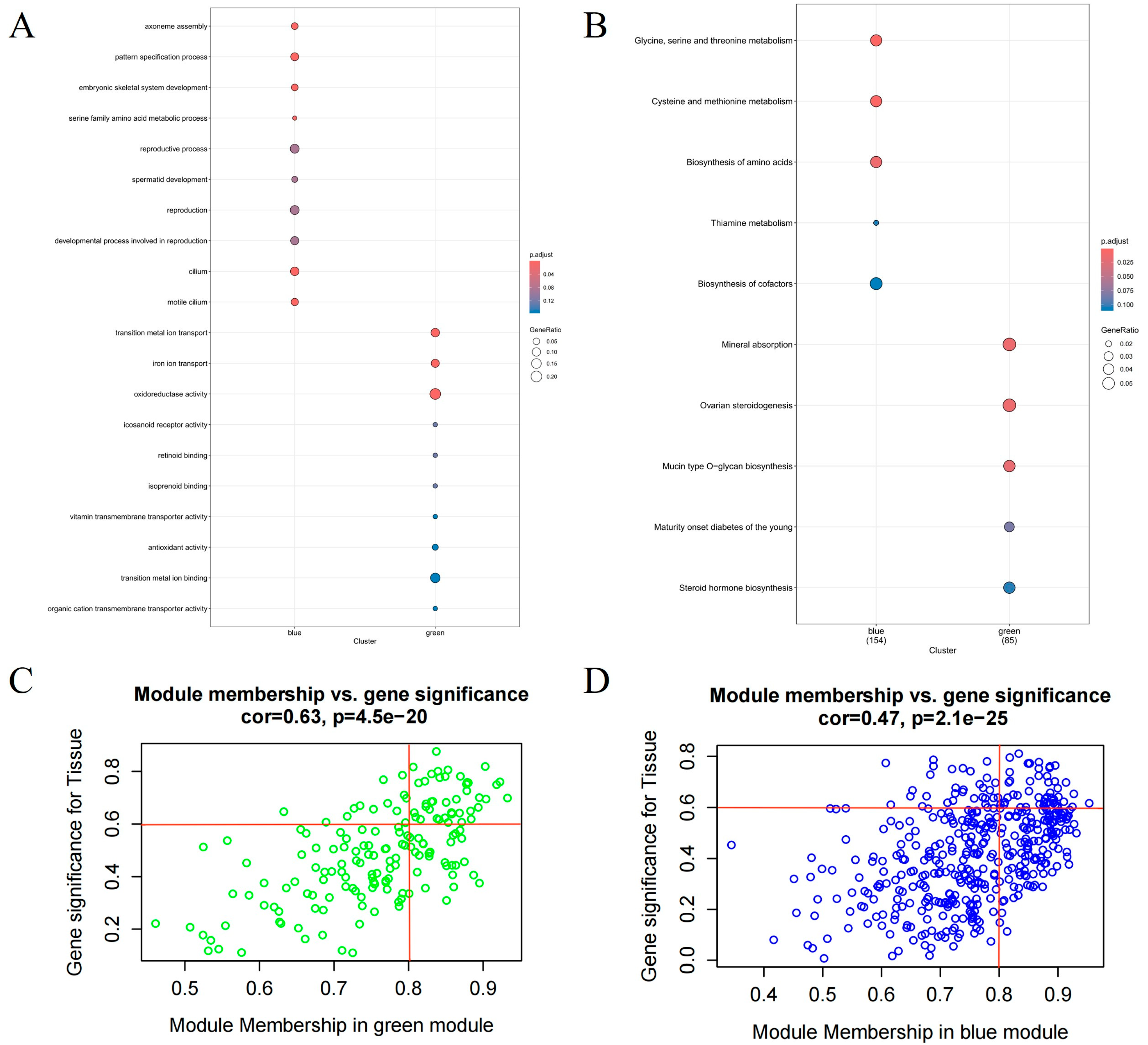
3.2. Screening of Hub Genes in Endometrium by WGCNA
3.3. Integration of eQTL Analysis and WGCNA Results for Identifying Candidate Genes and cis-eQTLs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benjamin, M.; Yik, S. Precision Livestock Farming in Swine Welfare: A Review for Swine Practitioners. Animals 2019, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Nonneman, D.J.; Lents, C.A. Functional Genomics of Reproduction in Pigs: Are We There Yet? Mol. Reprod. Dev. 2023, 90, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Zak, L.J.; Gaustad, A.H.; Bolarin, A.; Broekhuijse, M.L.W.J.; Walling, G.A.; Knol, E.F. Genetic Control of Complex Traits, with a Focus on Reproduction in Pigs. Mol. Reprod. Dev. 2017, 84, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Spötter, A.; Distl, O. Genetic Approaches to the Improvement of Fertility Traits in the Pig. Vet. J. 2006, 172, 234–247. [Google Scholar] [CrossRef]
- Yin, C.; Zhou, P.; Wang, Y.; Yin, Z.; Liu, Y. Using Genomic Selection to Improve the Accuracy of Genomic Prediction for Multi-Populations in Pigs. Animal 2024, 18, 101062. [Google Scholar] [CrossRef]
- Almeida, F.R.C.L.; Dias, A.L.N.A. Pregnancy in Pigs: The Journey of an Early Life. Domest. Anim. Endocrinol. 2022, 78, 106656. [Google Scholar] [CrossRef]
- Kridli, R.T.; Khalaj, K.; Bidarimath, M.; Tayade, C. Placentation, Maternal–Fetal Interface, and Conceptus Loss in Swine. Theriogenology 2016, 85, 135–144. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Z.; Wang, J.; Zeng, T.; Ai, X.; Wu, K. An Integrative ATAC-Seq and RNA-Seq Analysis of the Endometrial Tissues of Meishan and Duroc Pigs. Int. J. Mol. Sci. 2023, 24, 14812. [Google Scholar] [CrossRef]
- Li, W.-T.; Zhang, M.-M.; Li, Q.-G.; Tang, H.; Zhang, L.-F.; Wang, K.-J.; Zhu, M.-Z.; Lu, Y.-F.; Bao, H.-G.; Zhang, Y.-M.; et al. Whole-Genome Resequencing Reveals Candidate Mutations for Pig Prolificacy. Proc. R. Soc. B Biol. Sci. 2017, 284, 20172437. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, R.; Zhang, H.; Wang, D.; Wang, J.; Wu, K. A Unique 15-Bp InDel in the First Intron of BMPR1B Regulates Its Expression in Taihu Pigs. BMC Genom. 2022, 23, 799. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, H.; Wang, J.; Wang, D.; Zeng, T.; Ai, X.; Wang, X.; Zhao, X.; Wu, K. Functional Effects of BMPR1B in Porcine Endometrium Provides Novel Insights into the High Fecundity of Taihu Pigs. Int. J. Biol. Macromol. 2025, 293, 139188. [Google Scholar] [CrossRef]
- Xing, L.; Lu, X.; Zhang, W.; Wang, Q.; Zhang, W. Genetic Structure and Genome-Wide Association Analysis of Growth and Reproductive Traits in Fengjing Pigs. Animals 2024, 14, 2449. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.X.; Gao, G.X.; Zhou, Y.; Guo, C.X.; Li, B.; El-Ashram, S.; Li, Z.L. Genome-Wide Association Studies Uncover Genes Associated with Litter Traits in the Pig. Animal 2022, 16, 100672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, Z.; Ye, S.; He, Y.; Huang, S.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J. Genome-Wide Association Study for Reproductive Traits in a Duroc Pig Population. Animals 2019, 9, 732. [Google Scholar] [CrossRef]
- Schipper, M.; Posthuma, D. Demystifying Non-Coding GWAS Variants: An Overview of Computational Tools and Methods. Hum. Mol. Genet. 2022, 31, R73–R83. [Google Scholar] [CrossRef]
- Flynn, E.D.; Lappalainen, T. Functional Characterization of Genetic Variant Effects on Expression. Annu. Rev. Biomed. Data Sci. 2022, 5, 119–139. [Google Scholar] [CrossRef]
- Pai, A.A.; Pritchard, J.K.; Gilad, Y. The Genetic and Mechanistic Basis for Variation in Gene Regulation. PLoS Genet. 2015, 11, e1004857. [Google Scholar] [CrossRef]
- Safaei, S.M.H.; Dadpasand, M.; Mohammadabadi, M.; Atashi, H.; Stavetska, R.; Klopenko, N.; Kalashnyk, O. An Origanum majorana Leaf Diet Influences Myogenin Gene Expression, Performance, and Carcass Characteristics in Lambs. Animals 2023, 13, 14. [Google Scholar] [CrossRef]
- Mohammadabadi, M.R.; Torabi, A.; Tahmourespoor, M.; Baghizadeh, A.; Koshkoie, A.E.; Mohammadi, A. Analysis of Bovine Growth Hormone Gene Polymorphism of Local and Holstein Cattle Breeds in Kerman Province of Iran Using Polymerase Chain Reaction Restriction Fragment Length Polymorphism (PCR-RFLP). Afr. J. Biotechnol. 2010, 9, 6848–6852. [Google Scholar]
- Cai, W.; Hu, J.; Zhang, Y.; Guo, Z.; Zhou, Z.; Hou, S. Cis-eQTLs in Seven Duck Tissues Identify Novel Candidate Genes for Growth and Carcass Traits. BMC Genom. 2024, 25, 429. [Google Scholar] [CrossRef]
- Cai, W.; Zhang, Y.; Chang, T.; Wang, Z.; Zhu, B.; Chen, Y.; Gao, X.; Xu, L.; Zhang, L.; Gao, H.; et al. The eQTL Colocalization and Transcriptome-Wide Association Study Identify Potentially Causal Genes Responsible for Economic Traits in Simmental Beef Cattle. J. Anim. Sci. Biotechnol. 2023, 14, 78. [Google Scholar] [CrossRef]
- Drag, M.H.; Kogelman, L.J.A.; Maribo, H.; Meinert, L.; Thomsen, P.D.; Kadarmideen, H.N. Characterization of eQTLs Associated with Androstenone by RNA Sequencing in Porcine Testis. Physiol. Genom. 2019, 51, 488–499. [Google Scholar] [CrossRef] [PubMed]
- The GTEx Consortium; Aguet, F.; Anand, S.; Ardlie, K.G.; Gabriel, S.; Getz, G.A.; Graubert, A.; Hadley, K.; Handsaker, R.E.; Huang, K.H.; et al. The GTEx Consortium Atlas of Genetic Regulatory Effects across Human Tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Teng, J.; Gao, Y.; Yin, H.; Bai, Z.; Liu, S.; Zeng, H.; Bai, L.; Cai, Z.; Zhao, B.; Li, X.; et al. A Compendium of Genetic Regulatory Effects across Pig Tissues. Nat. Genet. 2024, 56, 112–123. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Stegle, O.; Parts, L.; Piipari, M.; Winn, J.; Durbin, R. Using Probabilistic Estimation of Expression Residuals (PEER) to Obtain Increased Power and Interpretability of Gene Expression Analyses. Nat. Protoc. 2012, 7, 500–507. [Google Scholar] [CrossRef]
- Taylor-Weiner, A.; Aguet, F.; Haradhvala, N.J.; Gosai, S.; Anand, S.; Kim, J.; Ardlie, K.; Van Allen, E.M.; Getz, G. Scaling Computational Genomics to Millions of Individuals with GPUs. Genome Biol. 2019, 20, 228. [Google Scholar] [CrossRef]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the Future: Meeting New Challenges and Providing Updated Services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jiang, W.; Zhao, H. Integration of Expression QTLs with Fine Mapping via SuSiE. PLoS Genet. 2024, 20, e1010929. [Google Scholar] [CrossRef]
- Mohammadi, P.; Castel, S.E.; Brown, A.A.; Lappalainen, T. Quantifying the Regulatory Effect Size of Cis-Acting Genetic Variation Using Allelic Fold Change. Genome Res. 2017, 27, 1872. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Xing, K.; Liu, H.; Zhang, F.; Liu, Y.; Shi, Y.; Ding, X.; Wang, C. Identification of Key Genes Affecting Porcine Fat Deposition Based on Co-Expression Network Analysis of Weighted Genes. J. Anim. Sci. Biotechnol. 2021, 12, 100. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Guan, D.; Bai, Z.; Zhu, X.; Zhong, C.; Hou, Y.; Lan, F.; Diao, S.; Yao, Y.; Zhao, B.; Zhu, D.; et al. The ChickenGTEx Pilot Analysis: A Reference of Regulatory Variants across 28 Chicken Tissues. bioRxiv 2023. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, J.; Li, W.; Liu, X.; Chen, S.; Mi, S.; Yang, J.; Teng, J.; Fang, L.; Yu, Y. Identification and Characterization of Whole Blood Gene Expression and Splicing Quantitative Trait Loci during Early to Mid-Lactation of Dairy Cattle. BMC Genom. 2024, 25, 445. [Google Scholar] [CrossRef]
- Diniz, W.J.S.; Afonso, J.; Kertz, N.C.; Dyce, P.W.; Banerjee, P. Mapping Expression Quantitative Trait Loci Targeting Candidate Genes for Pregnancy in Beef Cows. Biomolecules 2024, 14, 150. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and Limitations of Genome-Wide Association Studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Aguet, F.; Alasoo, K.; Li, Y.I.; Battle, A.; Im, H.K.; Montgomery, S.B.; Lappalainen, T. Molecular Quantitative Trait Loci. Nat. Rev. Methods Primers 2023, 3, 4. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-Scale Cis- and Trans-eQTL Analyses Identify Thousands of Genetic Loci and Polygenic Scores That Regulate Blood Gene Expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Schaid, D.J.; Chen, W.; Larson, N.B. From Genome-Wide Associations to Candidate Causal Variants by Statistical Fine-Mapping. Nat. Rev. Genet. 2018, 19, 491. [Google Scholar] [CrossRef]
- Kim, J.-M.; Park, J.-E.; Yoo, I.; Han, J.; Kim, N.; Lim, W.-J.; Cho, E.-S.; Choi, B.; Choi, S.; Kim, T.-H.; et al. Integrated Transcriptomes throughout Swine Oestrous Cycle Reveal Dynamic Changes in Reproductive Tissues Interacting Networks. Sci. Rep. 2018, 8, 5436. [Google Scholar] [CrossRef]
- Forde, N.; Simintiras, C.A.; Sturmey, R.; Mamo, S.; Kelly, A.K.; Spencer, T.E.; Bazer, F.W.; Lonergan, P. Amino Acids in the Uterine Luminal Fluid Reflects the Temporal Changes in Transporter Expression in the Endometrium and Conceptus during Early Pregnancy in Cattle. PLoS ONE 2014, 9, e100010. [Google Scholar] [CrossRef]
- Jia, Z.; Wei, Y.; Zhang, Y.; Song, K.; Yuan, J. Metabolic Reprogramming and Heterogeneity during the Decidualization Process of Endometrial Stromal Cells. Cell Commun. Signal. 2024, 22, 385. [Google Scholar] [CrossRef]
- Yang, T.; Zhao, J.; Liu, F.; Li, Y. Lipid Metabolism and Endometrial Receptivity. Hum. Reprod. Update 2022, 28, 858–889. [Google Scholar] [CrossRef]
- Artimovič, P.; Badovská, Z.; Toporcerová, S.; Špaková, I.; Smolko, L.; Sabolová, G.; Kriváková, E.; Rabajdová, M. Oxidative Stress and the Nrf2/PPARγ Axis in the Endometrium: Insights into Female Fertility. Cells 2024, 13, 1081. [Google Scholar] [CrossRef]
- Vaishnav, S.; Chauhan, A.; Ajay, A.; Saini, B.L.; Kumar, S.; Kumar, A.; Bhushan, B.; Gaur, G.K. Allelic to Genome Wide Perspectives of Swine Genetic Variation to Litter Size and Its Component Traits. Mol. Biol. Rep. 2023, 50, 3705–3721. [Google Scholar] [CrossRef]
- Nakamura, H.; Jasper, M.J.; Hull, M.L.; Aplin, J.D.; Robertson, S.A. Macrophages Regulate Expression of A1,2-Fucosyltransferase Genes in Human Endometrial Epithelial Cells. Mol. Hum. Reprod. 2012, 18, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Feng, H.; Cao, Y.; Huang, Y.; Dai, C.; Wu, S.; Bao, W. New Insight into the Molecular Mechanism of the FUT2 Regulating Escherichia coli F18 Resistance in Weaned Piglets. Int. J. Mol. Sci. 2018, 19, 3301. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, S.; Wu, J.; Sun, S.; Wu, S.; Bao, W. Association Analysis of the SNP (Rs345476947) in the FUT2 Gene with the Production and Reproductive Traits in Pigs. Genes Genom. 2018, 40, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xue, M.; Zhang, J.; Yu, H.; Gu, Y.; Du, M.; Ye, W.; Wan, B.; Jin, M.; Zhang, Y. Protective Role of GPR120 in the Maintenance of Pregnancy by Promoting Decidualization via Regulation of Glucose Metabolism. EBioMedicine 2019, 39, 540–551. [Google Scholar] [CrossRef]
- Shi, Q.; Song, X.; Wang, J.; Gu, J.; Zhang, W.; Hu, J.; Zhou, X.; Yu, R. FRK Inhibits Migration and Invasion of Human Glioma Cells by Promoting N-Cadherin/β-Catenin Complex Formation. J. Mol. Neurosci. 2015, 55, 32–41. [Google Scholar] [CrossRef]
- Tamate, M.; Tanaka, R.; Osogami, H.; Matsuura, M.; Satohisa, S.; Iwasaki, M.; Saito, T. Rap1GAP Inhibits Tumor Progression in Endometrial Cancer. Biochem. Biophys. Res. Commun. 2017, 485, 476–483. [Google Scholar] [CrossRef]
- D’Occhio, M.J.; Campanile, G.; Zicarelli, L.; Visintin, J.A.; Baruselli, P.S. Adhesion Molecules in Gamete Transport, Fertilization, Early Embryonic Development, and Implantation—Role in Establishing a Pregnancy in Cattle: A Review. Mol. Reprod. Dev. 2020, 87, 206–222. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, Z.; Gao, X.; Luo, P.; Jiang, X. ARMC Subfamily: Structures, Functions, Evolutions, Interactions, and Diseases. Front. Mol. Biosci. 2021, 8, 791597. [Google Scholar] [CrossRef]
- Patir, A.; Fraser, A.M.; Barnett, M.W.; McTeir, L.; Rainger, J.; Davey, M.G.; Freeman, T.C. The Transcriptional Signature Associated with Human Motile Cilia. Sci. Rep. 2020, 10, 10814. [Google Scholar] [CrossRef]
- Spassky, N.; Meunier, A. The Development and Functions of Multiciliated Epithelia. Nat. Rev. Mol. Cell Biol. 2017, 18, 423–436. [Google Scholar] [CrossRef]
- Ikawa, M.; Wada, I.; Kominami, K.; Watanabe, D.; Toshimori, K.; Nishimune, Y.; Okabe, M. The Putative Chaperone Calmegin Is Required for Sperm Fertility. Nature 1997, 387, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.-L.; Han, W.-Q.; Yang, F.-P.; Ji, K.-D.; Wang, J.-G.; Gao, P.-J.; He, G.; Wu, S.-N. Association of a SNP in SLC35F3 Gene with the Risk of Hypertension in a Chinese Han Population. Front. Genet. 2016, 7, 108. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Lim, W.; Jo, G.; Bazer, F.W.; Song, G. Estrogen Regulation of Phosphoserine Phosphatase during Regression and Recrudescence of Female Reproductive Organs. Gen. Comp. Endocrinol. 2015, 214, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Uno, Y.; Shimizu, M.; Ogawa, Y.; Makiguchi, M.; Kawaguchi, H.; Yamato, O.; Ishizuka, M.; Yamazaki, H. Molecular and Functional Characterization of Flavin-Containing Monooxygenases in Pigs, Dogs, and Cats. Biochem. Pharmacol. 2022, 202, 115125. [Google Scholar] [CrossRef]
- Miller, M.M.; James, R.A.; Richer, J.K.; Gordon, D.F.; Wood, W.M.; Horwitz, K.B. Progesterone Regulated Expression of Flavin-Containing Monooxygenase 5 by the B-Isoform of Progesterone Receptors: Implications for Tamoxifen Carcinogenicity. J. Clin. Endocrinol. Metab. 1997, 82, 2956–2961. [Google Scholar] [CrossRef]
- Zhong, C.; Liu, Z.; Qiao, X.; Kang, L.; Sun, Y.; Jiang, Y. Integrated Transcriptomic Analysis on Small Yellow Follicles Reveals That Sosondowah Ankyrin Repeat Domain Family Member A Inhibits Chicken Follicle Selection. Anim. Biosci. 2020, 34, 1–13. [Google Scholar] [CrossRef]
- Belitškin, D.; Munne, P.; Pant, S.M.; Anttila, J.M.; Suleymanova, I.; Belitškina, K.; Kirchhofer, D.; Janetka, J.; Käsper, T.; Jalil, S.; et al. Hepsin Promotes Breast Tumor Growth Signaling via the TGFβ-EGFR Axis. Mol. Oncol. 2024, 18, 547–561. [Google Scholar] [CrossRef]
- Ding, J.; Matsumiya, T.; Miki, Y.; Hayakari, R.; Shiba, Y.; Kawaguchi, S.; Seya, K.; Imaizumi, T. ER Export Signals Mediate Plasma Membrane Localization of Transmembrane Protein TMEM72. FEBS J. 2023, 290, 2636–2657. [Google Scholar] [CrossRef]
- Shen, C.-Y.; Chang, W.-H.; Chen, Y.-J.; Weng, C.-W.; Regmi, P.; Kier, M.K.K.; Su, K.-Y.; Chang, G.-C.; Chen, J.-S.; Chen, Y.-J.; et al. Tissue Proteogenomic Landscape Reveals the Role of Uncharacterized SEL1L3 in Progression and Immunotherapy Response in Lung Adenocarcinoma. J. Proteome Res. 2023, 22, 1056–1070. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Significant | Independent | Fine Mapped |
|---|---|---|---|
| eGene | 5632 | 5632 | 1420 |
| eQTL | 34,876 | 6288 | 1496 |
| eGene-eQTL | 56,660 | 6677 | 1549 |
| Ensemble ID | Gene | eQTL Position | RS Number | PIP | Pval_Nominal |
|---|---|---|---|---|---|
| ENSSSCG00000004434 | FRK | 1_81907491 | rs333994665 | 0.9962785 | 6.31 × 10−6 |
| ENSSSCG00000011078 | ARMC3 | 10_52338433 | rs319228529 | 0.93160963 | 1.01 × 10−4 |
| ENSSSCG00000010162 | SLC35F3 | 14_56740841 | rs338283666 | 0.955412 | 1.02 × 10−4 |
| ENSSSCG00000010162 | SLC35F3 | 14_57972209 | rs324855920 | 0.9711524 | 1.53 × 10−4 |
| ENSSSCG00000025787 | TMEM72 | 14_90784347 | rs335313886 | 0.97402775 | 8.00 × 10−4 |
| ENSSSCG00000010478 | FFAR4 | 14_104040504 | rs341695379 | 0.9788259 | 2.10 × 10−8 |
| ENSSSCG00000014286 | SOWAHA | 2_135149975 | rs81365495 | 0.9513308 | 1.41 × 10−6 |
| ENSSSCG00000007748 | PSPH | 3_16858682 | rs1109047347 | 0.97025245 | 1.75 × 10−7 |
| ENSSSCG00000006702 | FMO5 | 4_100371933 | rs336496720 | 0.9379143 | 5.29 × 10−5 |
| ENSSSCG00000021867 | HPN | 6_44606729 | rs692754439 | 0.9981295 | 4.59 × 10−10 |
| ENSSSCG00000003145 | FUT2 | 6_54046253 | rs323859419 | 0.97183824 | 1.16 × 10−4 |
| ENSSSCG00000003145 | FUT2 | 6_53069250 | rs327066479 | 0.97199005 | 2.44 × 10−4 |
| ENSSSCG00000022029 | RAP1GAP | 6_80017044 | rs55619266 | 0.9862745 | 2.21 × 10−6 |
| ENSSSCG00000001040 | C6orf52 | 7_7455876 | rs80974566 | 0.9836499 | 8.59 × 10−10 |
| ENSSSCG00000022446 | SEL1L3 | 8_19714479 | rs320263410 | 0.9991525 | 1.04 × 10−5 |
| ENSSSCG00000026360 | CLGN | 8_87641656 | rs343689578 | 0.98143643 | 1.69 × 10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, T.; Wang, J.; Liu, Z.; Wang, X.; Zhang, H.; Ai, X.; Deng, X.; Wu, K. Identification of Candidate Genes and eQTLs Related to Porcine Reproductive Function. Animals 2025, 15, 1038. https://doi.org/10.3390/ani15071038
Zeng T, Wang J, Liu Z, Wang X, Zhang H, Ai X, Deng X, Wu K. Identification of Candidate Genes and eQTLs Related to Porcine Reproductive Function. Animals. 2025; 15(7):1038. https://doi.org/10.3390/ani15071038
Chicago/Turabian StyleZeng, Tong, Ji Wang, Zhexi Liu, Xiaofeng Wang, Han Zhang, Xiaohua Ai, Xuemei Deng, and Keliang Wu. 2025. "Identification of Candidate Genes and eQTLs Related to Porcine Reproductive Function" Animals 15, no. 7: 1038. https://doi.org/10.3390/ani15071038
APA StyleZeng, T., Wang, J., Liu, Z., Wang, X., Zhang, H., Ai, X., Deng, X., & Wu, K. (2025). Identification of Candidate Genes and eQTLs Related to Porcine Reproductive Function. Animals, 15(7), 1038. https://doi.org/10.3390/ani15071038