Identification of Key Genes Related to Intramuscular Fat Content of Psoas Major Muscle in Saba Pigs by Integrating Bioinformatics and Machine Learning Based on Transcriptome Data

 ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Preprocessing
2.2. Measurement of Fatty Acid and Amino Acid Composition in PMM
2.3. Detection of Differentially Expressed Genes (DEGs)
2.4. Functional Enrichment Analysis of DEGs
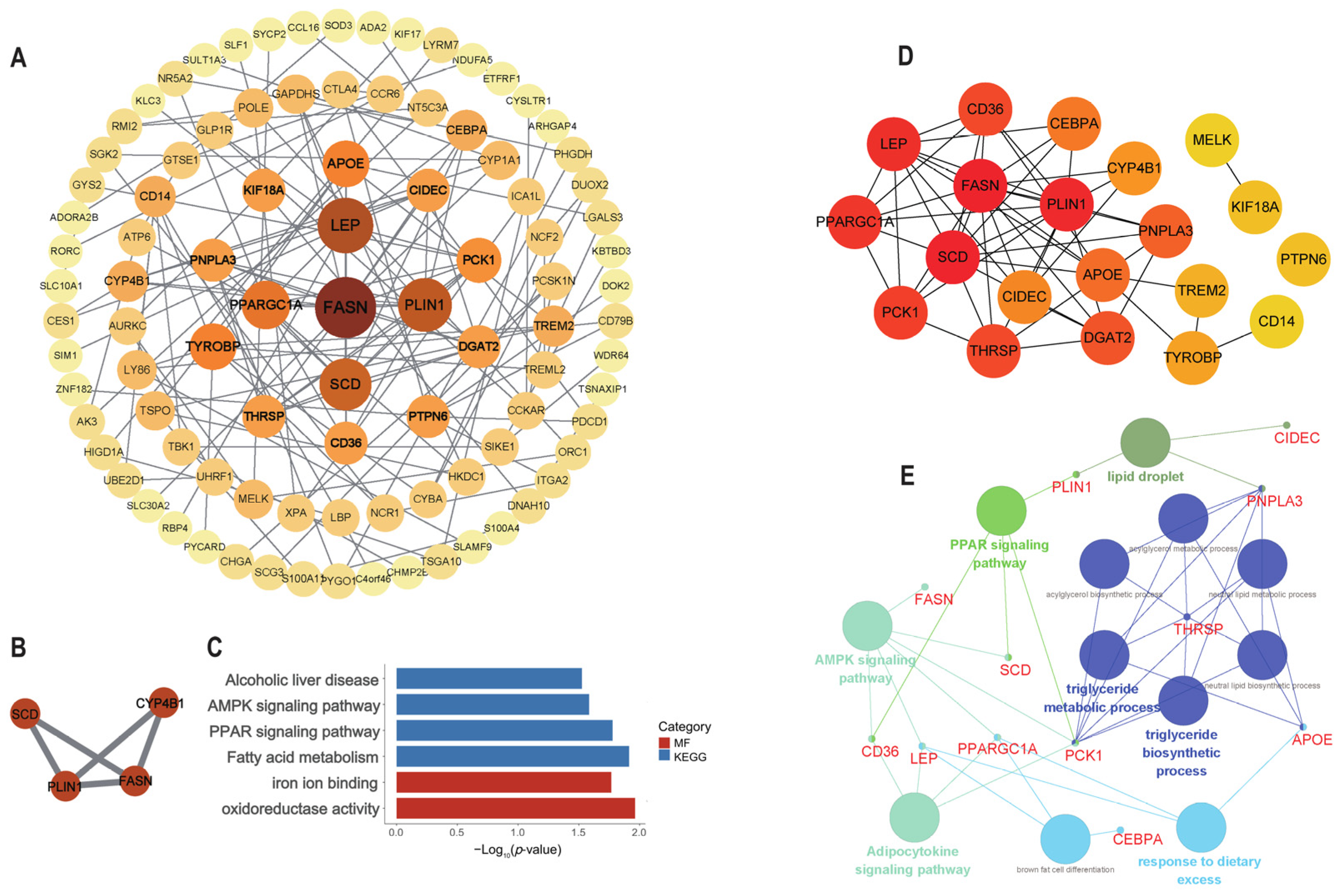
2.5. Protein–Protein Interaction (PPI) Network Analysis and Hub Gene Recognition
2.6. Screening of Potential IMF-Related Genes
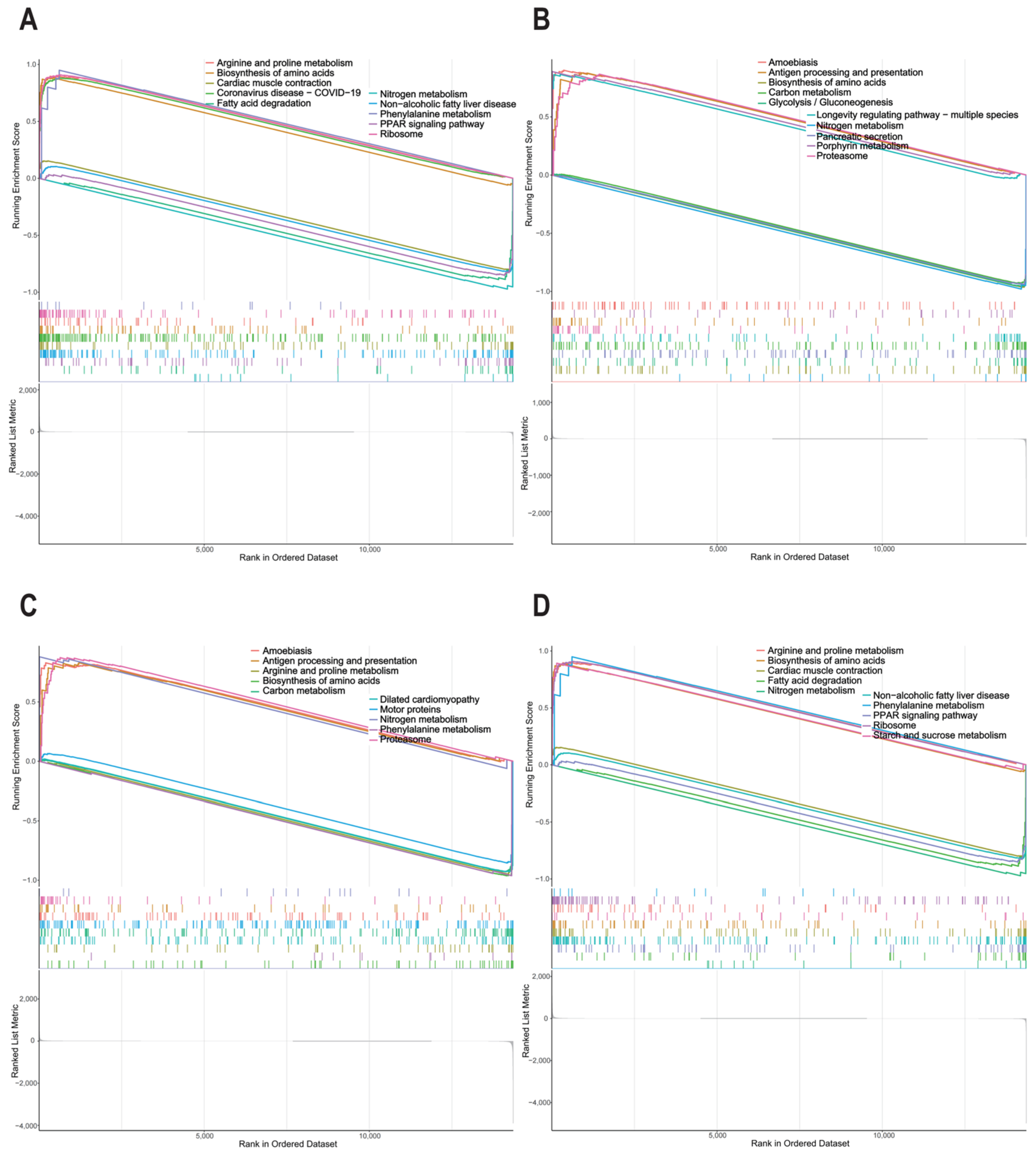
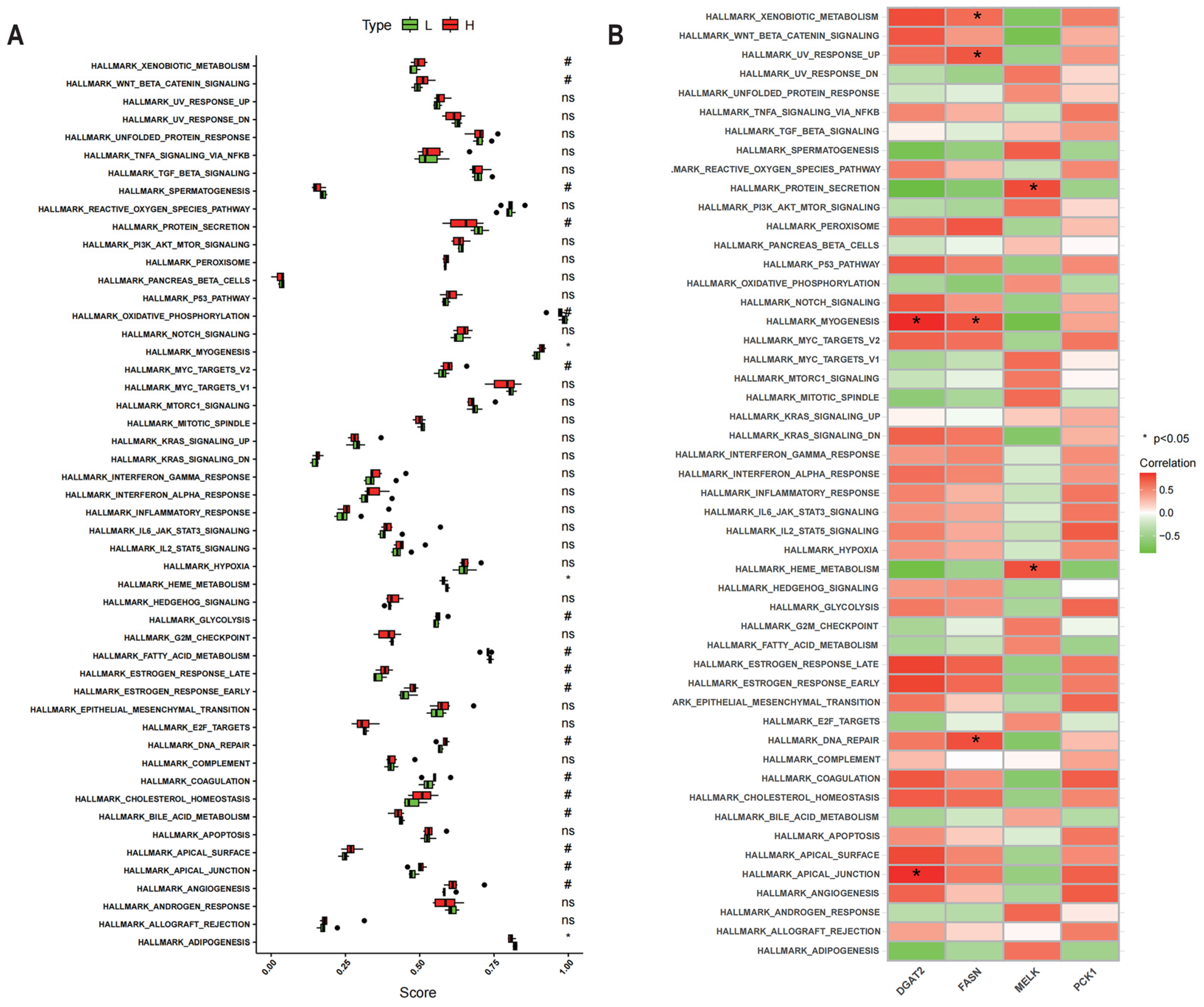
2.7. Gene Set Enrichment Analysis of Potential IMF-Related Genes
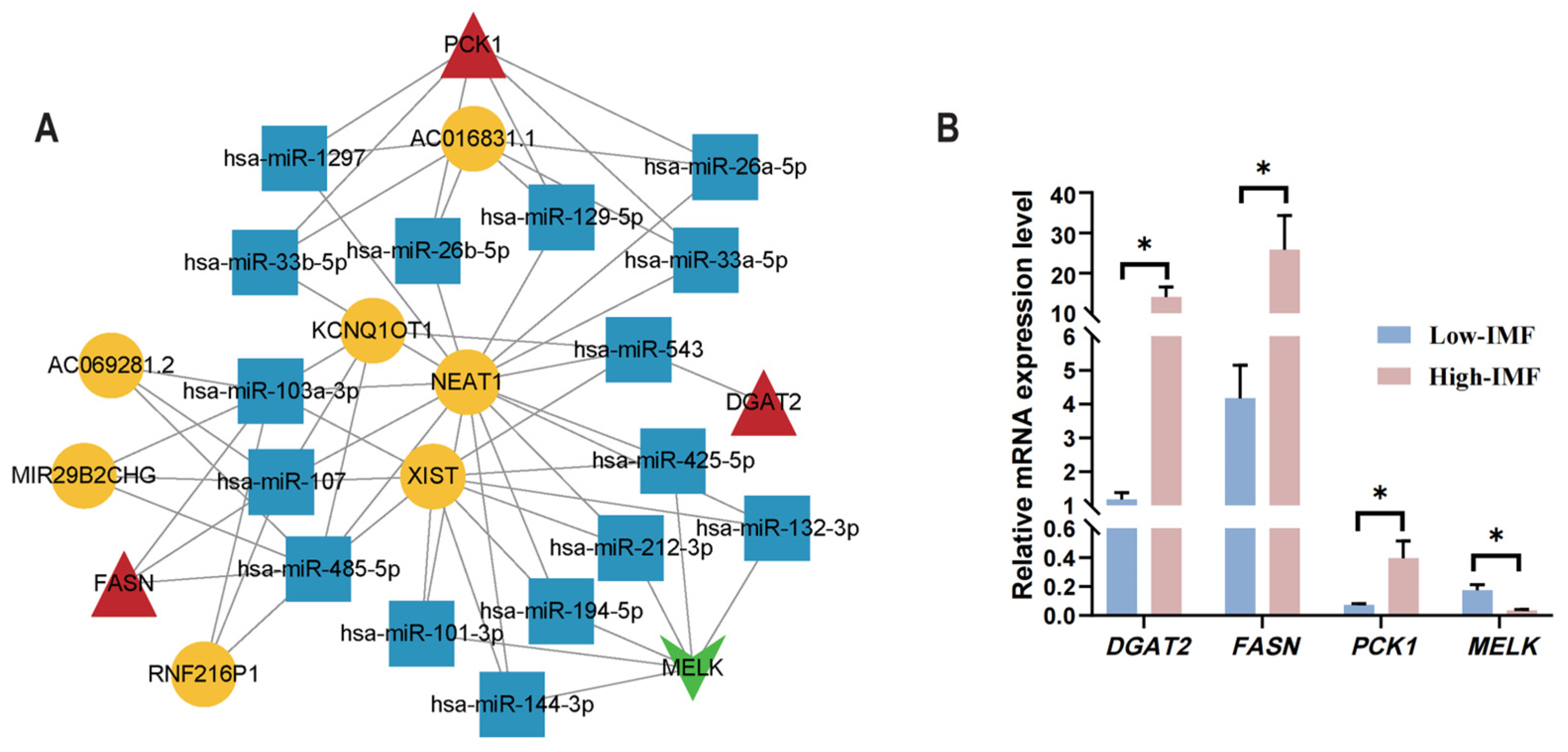
2.8. Construction of the mRNA (Gene)–miRNA–lncRNA Regulation Network
2.9. Verification of RNA Sequencing Results by RT-qPCR
2.10. Statistical Analysis
3. Results
3.1. The Fatty Acid and Amino Acid Composition of PMM Samples
3.2. DEG Identification
3.3. Functional Enrichment Analysis of DEGs
3.4. PPI Network Construction
3.5. Identification and Analysis of Hub Genes
3.6. Identification of Potential IMF-Related Genes
3.7. Analysis of Hallmark Gene Sets of Potential IMF-Related Genes
3.8. Development of the mRNA (Gene)–miRNA–lncRNA Interaction Network
3.9. Validation of the Potential Genes via RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhan, H.; Xiong, Y.; Wang, Z.; Dong, W.; Zhou, Q.; Xie, S.; Li, X.; Zhao, S.; Ma, Y. Integrative analysis of transcriptomic and metabolomic profiles reveal the complex molecular regulatory network of meat quality in Enshi black pigs. Meat Sci. 2022, 183, 108642. [Google Scholar] [CrossRef]
- Song, B.; Zheng, C.; Zheng, J.; Zhang, S.; Zhong, Y.; Guo, Q.; Li, F.; Long, C.; Xu, K.; Duan, Y.; et al. Comparisons of carcass traits, meat quality, and serum metabolome between haziling and Yorkshire pigs. Anim. Nutr. 2022, 8, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Jung, J.; Park, E.; Kim, H. Identification of genes related to intramuscular fat content of pigs using genome-wide association study. Asian-Australas. J. Anim. Sci. 2018, 31, 157–162. [Google Scholar] [CrossRef]
- Ding, R.; Yang, M.; Quan, J.; Li, S.; Zhuang, Z.; Zhou, S.; Zheng, E.; Hong, L.; Li, Z.; Cai, G.; et al. Single-locus and multi-locus genome-wide association studies for intramuscular fat in Duroc pigs. Front. Genet. 2019, 10, 619. [Google Scholar] [CrossRef] [PubMed]
- Wojtysiak, D.; Gorska, M.; Wojciechowska, J. Muscle fibre characteristics and physico-chemical parameters of m. semimembranosus from Puławska, Polish Large White and Pietrain pigs. Folia Biol. 2016, 64, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Long, H.; Feng, S.; Ma, T.; Wang, M.; Niu, L.; Zhang, X.; Wang, L.; Lei, Y.; Chen, Y.; et al. Trait correlated expression combined with eQTL and ASE analyses identified novel candidate genes affecting intramuscular fat. BMC Genom. 2021, 22, 805. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Zhang, L.; Wang, L.; He, J.; Ma, H.; Wang, L. Preliminary identification and analysis of differential RNA editing between higher and lower backfat thickness pigs using DNA-seq and RNA-seq data. Anim. Genet. 2022, 53, 327–339. [Google Scholar] [CrossRef]
- Wang, L.; Xie, Y.; Chen, W.; Zhang, Y.; Zeng, Y. Identification and functional prediction of long noncoding RNAs related to intramuscular fat content in Laiwu pigs. Anim. Biosci. 2021, 35, 115. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, S.; Liu, H.; Ren, R.; Dong, Q.; Xie, J.; Cao, J. Profiling and characterization of miRNAs associated with intramuscular fat content in Yorkshire pigs. Anim. Biotechnol. 2020, 31, 256–263. [Google Scholar] [CrossRef]
- Han, Q.; Huang, X.; He, J.; Zeng, Y.; Yin, J.; Yin, Y. Intramuscular fat deposition in pig: A key target for improving pork quality. J. Integr. Agric. 2024; in press. [Google Scholar] [CrossRef]
- Li, Q.; Huang, Z.; Zhao, W.; Li, M.; Li, C. Transcriptome analysis reveals long intergenic non-coding RNAs contributed to intramuscular fat content differences between Yorkshire and Wei pigs. Int. J. Mol. Sci. 2020, 21, 1732. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Hou, Y.; Ling, Z.; Chen, Q.; Xu, T.; Liu, L.; Yu, N.; Ni, W.; Ding, X.; Zhang, X.; et al. Identification of candidate genes and regulatory competitive endogenous RNA (ceRNA) networks underlying intramuscular fat content in Yorkshire pigs with extreme fat deposition phenotypes. Int. J. Mol. Sci. 2022, 23, 12596. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Li, M.; Yan, D.; Ge, C. Germplasm characteristics, conservation and various utilization of Yunnan local pig breeds. J. Yunnan Agric. Univ. (Nat. Sci.) 2020, 35, 1096–1105. [Google Scholar] [CrossRef]
- Picard, B.; Gagaoua, M. Muscle fiber properties in cattle and their relationships with meat qualities: An overview. J. Agric. Food Chem. 2020, 68, 6021–6039. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cao, G.; Gao, P.; Jia, G.; Yang, F.; Meng, J. Comparing the mRNA expression profile of psoas major and longissimus dorsi muscles in pig. Indian J. Anim. Res. 2020, 54, 1490–1496. [Google Scholar] [CrossRef]
- Velotto, S.; Vitale, C.; Varricchio, E.; Crasto, A. A new perspective: An Italian autochthonous pig and its muscle and fat tissue characteristics. Indian J. Anim. Res. 2014, 48, 143–149. [Google Scholar] [CrossRef]
- Liu, Y.; Li, M.; Ma, J.; Zhang, J.; Zhou, C.; Wang, T.; Gao, X.; Li, X. Identification of differences in microRNA transcriptomes between porcine oxidative and glycolytic skeletal muscles. BMC Mol. Biol. 2013, 14, 7. [Google Scholar] [CrossRef]
- Wang, X.; Shi, S.; Wang, G.; Luo, W.; Wei, X.; Qiu, A.; Luo, F.; Ding, X. Using machine learning to improve the accuracy of genomic prediction of reproduction traits in pigs. J. Anim. Sci. Biotechnol. 2022, 13, 60. [Google Scholar] [CrossRef]
- Maltecca, C.; Lu, D.; Schillebeeckx, C.; McNulty, N.P.; Schwab, C.; Shull, C.; Tiezzi, F. Predicting growth and carcass traits in swine using microbiome data and machine learning algorithms. Sci. Rep. 2019, 9, 6574. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, X.; Wang, S.; Chen, Q.; Li, M.; Lu, S. Identification of potential sex-specific biomarkers in pigs with low and high intramuscular fat content using integrated bioinformatics and machine learning. Genes 2023, 14, 1695. [Google Scholar] [CrossRef]
- Ge, F.; Li, J.; Gao, H.; Wang, X.; Zhang, X.; Gao, H.; Zhang, L.; Xu, L.; Gao, X.; Zhu, B.; et al. Comparative analysis of carcass traits and meat quality in indigenous Chinese cattle breeds. J. Food Compos. Anal. 2023, 124, 105645. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for visualization and analysis of biological networks. J. Data Min. Genom. Proteom. 2011, 18, 291–303. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinf. 2003, 4, 2. [Google Scholar] [CrossRef]
- Chin, C.; Chen, S.; Wu, H.; Ho, C.; Ko, M.; Lin, C. CytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010, 33, 21–22. [Google Scholar] [CrossRef]
- Rigatti, S.J. Random forest. J. Insur. Med. 2017, 47, 31–39. [Google Scholar] [CrossRef]
- Acosta, J.; Li, Q.; Freeburg, N.F.; Murali, N.; Indeglia, A.; Grothusen, G.P.; Cicchini, M.; Mai, H.; Gladstein, A.C.; Adler, K.M.; et al. p53 restoration in small cell lung cancer identifies a latent cyclophilin-dependent necrosis mechanism. Nat. Commun. 2023, 14, 4403. [Google Scholar] [CrossRef]
- Wang, L.; Wang, L.; He, P. Comprehensive analysis of immune-related gene signature based on ssGSEA algorithms in the prognosis and immune landscape of hepatocellular carcinoma. Front. Genet. 2022, 13, 1064432. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The rosetta stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human microRNA targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Ren, B.; Wang, H.; Ren, L.; Yangdan, C.; Zhou, Y.; Fan, H.; Lv, Y. Screening for microRNA-based diagnostic markers in hepatic alveolar echinococcosis. Medicine 2019, 98, e17156. [Google Scholar] [CrossRef]
- Vejnar, C.E.; Zdobnov, E.M. MiRmap: Comprehensive prediction of microRNA target repression strength. Nucleic Acids Res. 2012, 40, 11673–11683. [Google Scholar] [CrossRef]
- Martins, J.M.; Neves, J.A.; Freitas, A.; Tirapicos, J.L. Rearing system and oleic acid supplementation effect on carcass and lipid characteristics of two muscles from an obese pig breed. Animal 2015, 9, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tang, Q.; Fan, L.; Xu, X.; Song, Z.; Hayat, K.; Feng, T.; Wang, Y. Identification of pork flavour precursors from enzyme-treated lard using Maillard model system assessed by GC–MS and partial least squares regression. Meat Sci. 2017, 124, 15–24. [Google Scholar] [CrossRef]
- Cameron, N.D.; Enser, M.; Nute, G.R.; Whittington, F.M.; Penman, J.C.; Fisken, A.C.; Perry, A.M.; Wood, J.D. Genotype with nutrition interaction on fatty acid composition of intramuscular fat and the relationship with flavour of pig meat. Meat Sci. 2000, 55, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Picard, B.; Lebret, B.; Cassar-Malek, I.; Liaubet, L.; Berri, C.; Le Bihan-Duval, E.; Hocquette, J.F.; Renand, G. Recent advances in omic technologies for meat quality management. Meat Sci. 2015, 109, 18–26. [Google Scholar] [CrossRef] [PubMed]
- De, S.S.; Raes, K.; Demeyer, D. Meat fatty acid composition as affected by fatness and genetic factors: A review. Anim. Res. 2004, 53, 81–98. [Google Scholar] [CrossRef]
- Luan, A.; Paik, K.J.; Li, J.; Zielins, E.R.; Atashroo, D.A.; Spencley, A.; Momeni, A.; Longaker, M.T.; Wang, K.C.; Wan, D.C. RNA sequencing for identification of differentially expressed noncoding transcripts during adipogenic differentiation of adipose-derived stromal cells. Plast. Reconstr. Surg. 2015, 136, 752–763. [Google Scholar] [CrossRef]
- Zhang, X.; Gu, S.; Shen, S.; Luo, T.; Zhao, H.; Liu, S.; Feng, J.; Yang, M.; Yi, L.; Fan, Z.; et al. Identification of circular RNA profiles in the liver of diet-induced obese mice and construction of the ceRNA network. Genes 2023, 14, 688. [Google Scholar] [CrossRef]
- Yu, X.X.; Murray, S.F.; Pandey, S.K.; Booten, S.L.; Bao, D.; Song, X.; Kelly, S.; Chen, S.; McKay, R.; Monia, B.P.; et al. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology 2005, 42, 362–371. [Google Scholar] [CrossRef]
- Qi, J.; Lang, W.; Giardino, E.; Caldwell, G.W.; Smith, C.; Minor, L.K.; Darrow, A.L.; Willemsens, G.; DeWaepenaert, K.; Roevens, P. High-content assays for evaluating cellular and hepatic diacylglycerol acyltransferase activity. J. Lipid Res. 2010, 51, 3559–3567. [Google Scholar] [CrossRef]
- Cui, J.; Zeng, Y.; Wang, H.; Chen, W.; Du, J.; Chen, Q.; Hu, Y.; Yang, L. The effects of DGAT1 and DGAT2 mRNA expression on fat deposition in fatty and lean breeds of pig. Livest. Sci. 2011, 140, 292–296. [Google Scholar] [CrossRef]
- Zang, L.; Wang, Y.; Sun, B.; Zhang, X.; Yang, C.; Kang, L.; Zhao, Z.; Jiang, Y. Identification of a 13bp indel polymorphism in the 3′-UTR of DGAT2 gene associated with backfat thickness and lean percentage in pigs. Gene 2016, 576, 729–733. [Google Scholar] [CrossRef]
- Jeong, J.; Kwon, E.G.; Im, S.K.; Seo, K.S.; Baik, M. Expression of fat deposition and fat removal genes is associated with intramuscular fat content in longissimus dorsi muscle of Korean cattle steers. J. Anim. Sci. 2012, 90, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Yin, Z.; Wang, M.; Zhang, W.; Raza, S.H.A.; Althobaiti, F.; Qi, L.; Wang, J. Expression of DGAT2 gene and its associations with intramuscular fat content and breast muscle fiber characteristics in domestic pigeons (Columba livia). Front. Vet. Sci. 2022, 9, 847363. [Google Scholar] [CrossRef]
- Zhang, J.; Choi, S.; Li, Q.; Wang, Y.; Sun, B.; Tang, L.; Wang, E.; Hua, H.; Li, X. Overexpression of DGAT2 stimulates lipid droplet formation and triacylglycerol accumulation in bovine satellite cells. Animals 2022, 12, 1847. [Google Scholar] [CrossRef]
- Guo, P.; Jin, X.; Zhang, J.; Li, Q.; Yan, C.; Li, X. Overexpression of DGAT2 regulates the differentiation of bovine preadipocytes. Animals 2023, 13, 1195. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kalhan, S.C.; Hanson, R.W. What is the metabolic role of phosphoenolpyruvate carboxykinase? Biol. Chem. 2009, 284, 27025–27029. [Google Scholar] [CrossRef] [PubMed]
- Semakova, J.; Hyroššová, P.; Méndez-Lucas, A.; Cutz, E.; Bermudez, J.; Burgess, S.; Alcántara, S.; Perales, J.C. PEPCK-C reexpression in the liver counters neonatal hypoglycemia in Pck1 del/del mice, unmasking role in non-gluconeogenic tissues. J. Physiol. Biochem. 2017, 73, 89–98. [Google Scholar] [CrossRef]
- Hakimi, P.; Yang, J.; Casadesus, G.; Massillon, D.; Tolentino-Silva, F.; Nye, C.K.; Cabrera, M.E.; Hagen, D.R.; Utter, C.B.; Baghdy, Y.; et al. Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) in skeletal muscle repatterns energy metabolism in the mouse. J. Biol. Chem. 2007, 282, 32844–32855. [Google Scholar] [CrossRef]
- Wang, W.; Xue, W.; Jin, B.; Zhang, X.; Ma, F.; Xu, X. Candidate gene expression affects intramuscular fat content and fatty acid composition in pigs. J. Appl. Genet. 2013, 54, 113–118. [Google Scholar] [CrossRef]
- Hudson, N.J.; Reverter, A.; Greenwood, P.L.; Guo, B.; Cafe, L.M.; Dalrymple, B.P. Longitudinal muscle gene expression patterns associated with differential intramuscular fat in cattle. Animal 2014, 9, 650–659. [Google Scholar] [CrossRef]
- Huang, J.; Feng, X.; Zhu, R.; Guo, D.; Wei, Y.; Cao, X.; Ma, Y.; Shi, D. Comparative transcriptome analysis reveals that PCK1 is a potential gene affecting IMF deposition in buffalo. BMC Genom. 2020, 21, 710. [Google Scholar] [CrossRef] [PubMed]
- Heyer, B.S.; Warsowe, J.; Solter, D.; Knowles, B.B.; Ackerman, S.L. New member of the Snf1/AMPK kinase family, Melk, is expressed in the mouse egg and preimplantation embryo. Mol. Reprod. Dev. 1997, 47, 148–156. [Google Scholar] [CrossRef]
- Mirey, G.; Chartrain, I.; Froment, C.; Quaranta, M.; Bouché, J.P.; Monsarrat, B.; Tassan, J.P.; Ducommun, B. CDC25B phosphorylated by pEg3 localizes to the centrosome and the spindle poles at mitosis. Cell Cycle 2005, 4, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Vulsteke, V.; Beullens, M.; Boudrez, A.; Keppens, S.; Van Eynde, A.; Rider, M.H.; Stalmans, W.; Bollen, M. Inhibition of spliceosome assembly by the cell cycle-regulated protein kinase MELK and involvement of splicing factor NIPP1. Biol. Chem. 2004, 279, 8642–8647. [Google Scholar] [CrossRef]
- Saito, R.; Tabata, Y.; Muto, A.; Arai, K.I.; Watanabe, S. Melk-like kinase plays a role in hematopoiesis in the zebra fish. Mol. Cell. Biol. 2005, 25, 6682–6693. [Google Scholar] [CrossRef] [PubMed]
- Nakano, I.; Paucar, A.A.; Bajpai, R.; Dougherty, J.D.; Zewail, A.; Kelly, T.K.; Kim, K.J.; Ou, J.; Groszer, M.; Imura, T.; et al. Maternal embryonic leucine zipper kinase (MELK) regulates multipotent neural progenitor proliferation. J. Cell. Biol. 2005, 170, 413–427. [Google Scholar] [CrossRef]
- Lin, M.L.; Park, J.H.; Nishidate, T.; Nakamura, Y.; Katagiri, T. Involvement of maternal embryonic leucine zipper kinase (MELK) in mammary carcinogenesis through interaction with Bcl-G, a pro-apoptotic member of the Bcl-2 family. Breast Cancer Res. 2007, 9, R17. [Google Scholar] [CrossRef]
- Seong, H.; Ha, H. Ablation of AMPK-related kinase MPK38/MELK leads to male-specific obesity in aged mature adult mice. Diabetes 2020, 70, 386–399. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Y.; Jiang, H.; Xiao, S.; Wang, S.; Ma, Q.; Sun, G.; Li, F.; Deng, Q.; Dai, L.; et al. Detection of differentially expressed genes in the longissimus dorsi of Northeastern Indigenous and Large White pigs. Genet. Mol. Res. 2011, 10, 779–791. [Google Scholar] [CrossRef]
- Zhao, S.; Ren, L.; Chen, L.; Zhang, X.; Cheng, M.; Li, W.; Zhang, Y.; Gao, S. Differential expression of lipid metabolism related genes in porcine muscle tissue leading to different intramuscular fat deposition. Lipids 2009, 44, 1029–1037. [Google Scholar] [CrossRef]
- Braglia, S.; Zappaterra, M.; Zambonelli, P.; Comella, M.; Dall’Olio, S.; Davoli, R. Analysis of g.265T>C SNP of fatty acid synthase gene and expression study in skeletal muscle and backfat tissues of Italian Large White and Italian Duroc pigs. Livest. Sci. 2014, 162, 15–22. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Q.; Chamba, Y.; Zhang, B.; Shang, P.; Zhang, H.; Wu, C. Identification of genes related to growth and lipid deposition from transcriptome profiles of pig muscle tissue. PLoS ONE 2015, 10, e0141138. [Google Scholar] [CrossRef]
- Muñoz, M.; García-Casco, J.M.; Caraballo, C.; Fernández-Barroso, M.Á.; Sánchez-Esquiliche, F.; Gómez, F.; Rodríguez, M.D.C.; Silió, L. Identification of candidate genes and regulatory factors underlying intramuscular fat content through longissimus dorsi transcriptome analyses in heavy Iberian pigs. Front. Genet. 2018, 9, 608. [Google Scholar] [CrossRef]
- Piao, M.Y.; Jo, C.; Kim, H.J.; Lee, H.J.; Kim, H.J.; Ko, J.Y.; Baik, M. Comparison of carcass and sensory traits and free amino acid contents among quality grades in loin and rump of Korean cattle steer. Asian-Australas. J. Anim. Sci. 2015, 28, 1629–1640. [Google Scholar] [CrossRef]
- He, K.; Wang, Q.; Wang, Z.; Pan, Y. Association study between gene polymorphisms in PPAR signaling pathway and porcine meat quality traits. Mamm. Genome 2013, 24, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Xia, C.; Cen, P.; Li, S.; Yu, L.; Zhu, J.; Jin, J. MiR-103-3p promotes hepatic steatosis to aggravate nonalcoholic fatty liver disease by targeting of ACOX1. Mol. Biol. Rep. 2022, 49, 7297–7305. [Google Scholar] [CrossRef]
- Bhatia, H.; Verma, G.; Datta, M. MiR-107 orchestrates ER stress induction and lipid accumulation by post-transcriptional regulation of fatty acid synthase in hepatocytes. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Mir, F.A.; Mall, R.; Iskandarani, A.; Ullah, E.; Samra, T.A.; Cyprian, F.; Parray, A.; Alkasem, M.; Abdalhakam, I.; Farooq, F.; et al. Characteristic microRNAs linked to dysregulated metabolic pathways in qatari adult subjects with obesity and metabolic syndrome. Front. Endocrinol. 2022, 13, 937089. [Google Scholar] [CrossRef]
- Wu, C.; Fang, S.; Zhang, H.; Li, X.; Du, Y.; Zhang, Y.; Lin, X.; Wang, L.; Ma, X.; Xue, Y.; et al. Long noncoding RNA XIST regulates brown preadipocytes differentiation and combats high-fat diet induced obesity by targeting C/EBPα. Mol. Med. 2022, 28, 6. [Google Scholar] [CrossRef]
- Surumi, B.; Jayakumaran Nair, A.; Rajalakshmi, R.; Prabha Kumari, C. Regulation of white adipogenesis by long non coding RNA NEAT1. Res. J. Biotechnol. 2022, 17, 11. [Google Scholar] [CrossRef]
- Shen, R.; Soeder, R.A.; Ophardt, H.D.; Giangrasso, P.J.; Cook, L.B. Identification of long non-coding RNAs expressed during early adipogenesis. OnLine J. Biol. Sci. 2019, 19, 245–259. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Number | Slaughter Age (d) | Live Weight Before Slaughter (kg) | IMF Content (%) |
|---|---|---|---|---|
| Low IMF | 6 | 320 | 102.67 ± 6.54 | 4.23 ± 0.61 B |
| High IMF | 6 | 320 | 100.58 ± 11.10 | 5.65 ± 0.78 A |
| Components | Low IMF | High IMF | p-Value |
|---|---|---|---|
| C14:0 | 0.042 ± 0.005 b | 0.052 ± 0.007 a | 0.022 |
| C16:0 | 0.938 ± 0.106 B | 1.778 ± 0.142 A | 0.008 |
| C17:0 | 0.007 ± 0.003 | 0.010 ± 0.003 | 0.179 |
| C18:0 | 0.593 ± 0.073 | 0.682 ± 0.083 | 0.078 |
| C20:0 | 0.014 ± 0.001 b | 0.017 ± 0.002 a | 0.017 |
| C23:0 | 0.001 ± 0.002 | 0.003 ± 0.002 | 0.069 |
| C24:0 | 0 | 0.003 ± 0.005 | 0.177 |
| C16:1 | 0.093 ± 0.013 B | 0.133 ± 0.026 A | 0.008 |
| C18:1n9c | 1.443 ± 0.188 B | 1.875 ± 0.260 A | 0.008 |
| C20:1 | 0.035 ± 0.007 | 0.051 ± 0.017 | 0.071 |
| C22:1n9 | 0.009 ± 0.005 | 0.006 ± 0.001 | 0.185 |
| C24:1 | 0.001 ± 0.001 | 0.003 ± 0.002 | 0.076 |
| C18:2n6c | 0.293 ± 0.043 | 0.346 ± 0.081 | 0.183 |
| C18:3n3 | 0.007 ± 0.001 | 0.008 ± 0.003 | 0.422 |
| C20:2 | 0.011 ± 0.002 b | 0.015 ± 0.003 a | 0.047 |
| C20:3n6 | 0.010 ± 0.001 | 0.011 ± 0.002 | 0.112 |
| C20:3n3 | 0 | 0.001 ± 0.002 | 0.175 |
| C20:4n6 | 0.061 ± 0.012 | 0.068 ± 0.012 | 0.318 |
| ΣSFA | 1.596 ± 0.179 b | 1.945 ± 0.233 a | 0.016 |
| ΣUFA | 1.963 ± 0.215 B | 2.516 ± 0.358 A | 0.009 |
| ΣMUFA | 1.581 ± 0.201 B | 2.067 ± 0.278 A | 0.007 |
| ΣPUFA | 0.382 ± 0.055 | 0.449 ± 0.091 | 0.149 |
| Σω-3 | 0.007 ± 0.001 | 0.009 ± 0.004 | 0.241 |
| Σω-6 | 0.364 ± 0.053 | 0.425 ± 0.085 | 0.160 |
| Σω-6/Σω-3 | 54.517 ± 4.027 | 51.707 ± 13.412 | 0.641 |
| Components | Low IMF | High IMF | p-Value |
|---|---|---|---|
| Aspartic (Asp) | 1.765 ± 0.076 | 1.750 ± 0.056 | 0.704 |
| Threonine (Thr) | 0.888 ± 0.033 | 0.887 ± 0.031 | 0.930 |
| Serine (Ser) | 0.698 ± 0.023 | 0.697 ± 0.022 | 0.900 |
| Glutamine (Glu) | 2.755 ± 0.119 | 2.732 ± 0.095 | 0.716 |
| Glycine (Gly) | 0.768 ± 0.029 | 0.773 ± 0.024 | 0.751 |
| Alanine (Ala) | 1.075 ± 0.035 | 1.073 ± 0.029 | 0.929 |
| Cysteine (Cys) | 0.185 ± 0.012 | 0.175 ± 0.019 | 0.299 |
| Valine (Val) | 0.973 ± 0.040 | 0.967 ± 0.026 | 0.738 |
| Methionine (Met) | 0.448 ± 0.031 | 0.438 ± 0.028 | 0.572 |
| Isoleucine (Ile) | 0.900 ± 0.043 | 0.888 ± 0.033 | 0.610 |
| Leucine (Leu) | 1.602 ± 0.063 | 1.602 ± 0.050 | 1.000 |
| Tyrosine (Tyr) | 0.605 ± 0.035 | 0.577 ± 0.029 | 0.161 |
| Phenylalanine (Phe) | 0.792 ± 0.026 | 0.787 ± 0.033 | 0.777 |
| Lysine (Lys) | 1.727 ± 0.063 | 1.722 ± 0.057 | 0.888 |
| Histidine (His) | 0.783 ± 0.053 | 0.758 ± 0.033 | 0.347 |
| Argine (Arg) | 1.190 ± 0.050 | 1.175 ± 0.037 | 0.568 |
| Proline (Pro) | 0.703 ± 0.020 | 0.723 ± 0.040 | 0.296 |
| FAA | 8.653 ± 0.324 | 8.635 ± 0.283 | 0.919 |
| EAA | 8.230 ± 0.335 | 8.178 ± 0.261 | 0.772 |
| NEAA | 10.528 ± 0.427 | 10.433 ± 0.308 | 0.668 |
| TAA | 17.858 ± 0.719 | 17.723 ± 0.526 | 0.720 |
| EAA/TAA | 0.461 ± 0.001 | 0.462 ± 0.002 | 0.573 |
| EAA/NEAA | 0.782 ± 0.003 | 0.784 ± 0.006 | 0.441 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Z.; Yang, Y.; Lai, J.; Chen, Q.; Wang, X.; Wang, S.; Li, M.; Lu, S. Identification of Key Genes Related to Intramuscular Fat Content of Psoas Major Muscle in Saba Pigs by Integrating Bioinformatics and Machine Learning Based on Transcriptome Data. Animals 2025, 15, 1181. https://doi.org/10.3390/ani15081181
Huang Z, Yang Y, Lai J, Chen Q, Wang X, Wang S, Li M, Lu S. Identification of Key Genes Related to Intramuscular Fat Content of Psoas Major Muscle in Saba Pigs by Integrating Bioinformatics and Machine Learning Based on Transcriptome Data. Animals. 2025; 15(8):1181. https://doi.org/10.3390/ani15081181
Chicago/Turabian StyleHuang, Zixia, Yongli Yang, Jinhua Lai, Qiang Chen, Xiaoyi Wang, Shuyan Wang, Mingli Li, and Shaoxiong Lu. 2025. "Identification of Key Genes Related to Intramuscular Fat Content of Psoas Major Muscle in Saba Pigs by Integrating Bioinformatics and Machine Learning Based on Transcriptome Data" Animals 15, no. 8: 1181. https://doi.org/10.3390/ani15081181
APA StyleHuang, Z., Yang, Y., Lai, J., Chen, Q., Wang, X., Wang, S., Li, M., & Lu, S. (2025). Identification of Key Genes Related to Intramuscular Fat Content of Psoas Major Muscle in Saba Pigs by Integrating Bioinformatics and Machine Learning Based on Transcriptome Data. Animals, 15(8), 1181. https://doi.org/10.3390/ani15081181