Design of the Crosslinking Reactions for Nucleic Acids-Binding Protein and Evaluation of the Reactivity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. General
2.2. Synthesis of the Nucleoside Derivatives
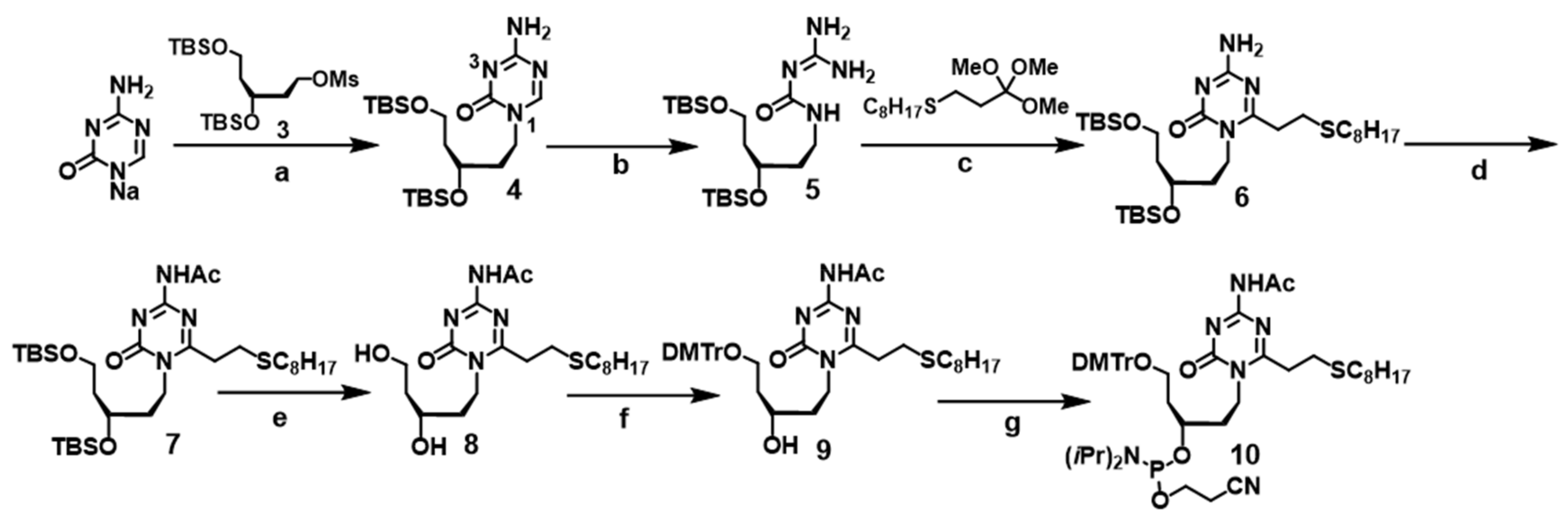
2.2.1. Synthesis of 4-amino-6-oxo-1-(3′,5′-O-t-butyldimethylsilyl-2′,4′-dideoxy-d-ribityl) triazine (4)
2.2.2. Synthesis of 4-amino-2-(2-octylthioethyl)-6-oxo-1-(3′,5′-O-t-butyldimethylsilyl-2′,4′-dideoxy-d-ribityl) triazine (6)
2.2.3. Synthesis of 2-(2-octylthioethyl)-6-oxo-4-acetylamino-1-(3′,5′-O-t-butyldimethylsilyl-2′,4′-dideoxy-d-ribityl) triazine (7)
2.2.4. Synthesis of 2-(2-octylthioethyl)-6-oxo-4-acetylamino-1-(2′,4′-dideoxy-d-ribityl) triazine (8)
2.2.5. Synthesis of 2-(2-octylthioethyl)-6-oxo-4-acetylamino-1-(5′-O-(4,4′-dimethoxytrityl)-2′,4′-dideoxy-d-ribityl) triazine (9)
2.2.6. Synthesis of 2-(2-octylthioethyl)-6-oxo-4-acetylamino-1-(3′-N,N-diisopropyl cyanoethyl-phosphoramidyl-5′-O-(4,4′-dimethoxytrityl)-2′,4′-dideoxy-d-ribityl) triazine (10)
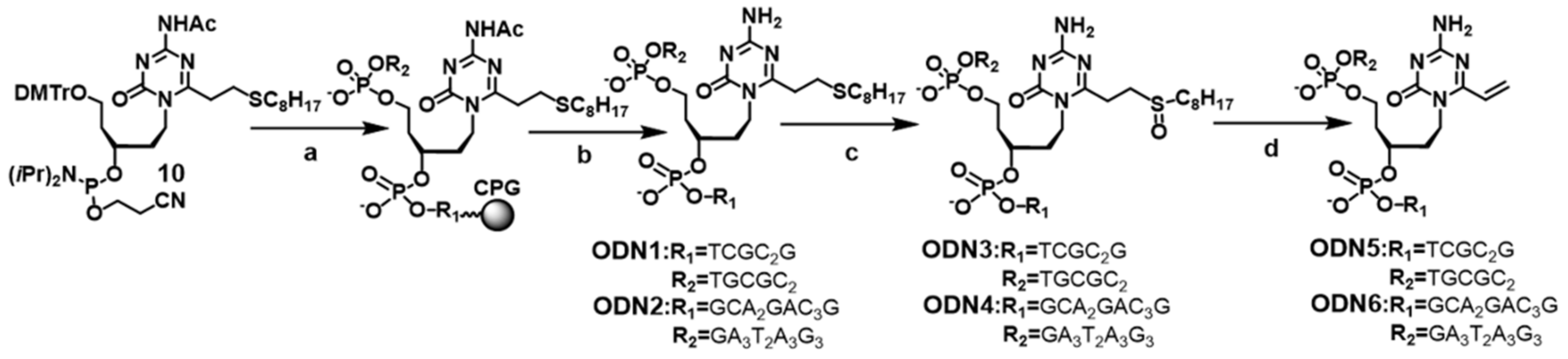
2.3. Synthesis of the Oligodeoxynucleotides Containing acyAOVT Derivatives
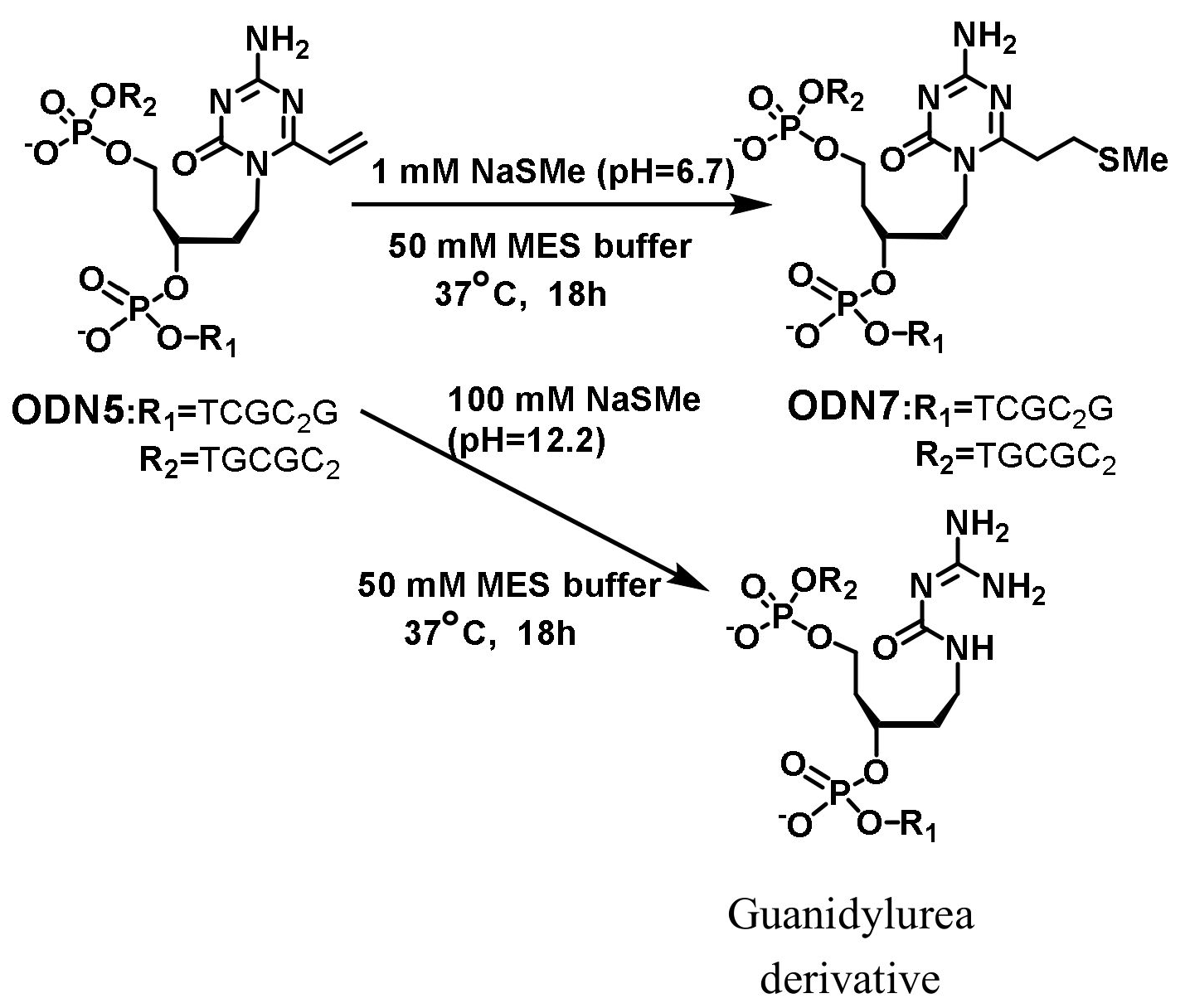
2.4. General Procedure for the Crosslinking Reactions
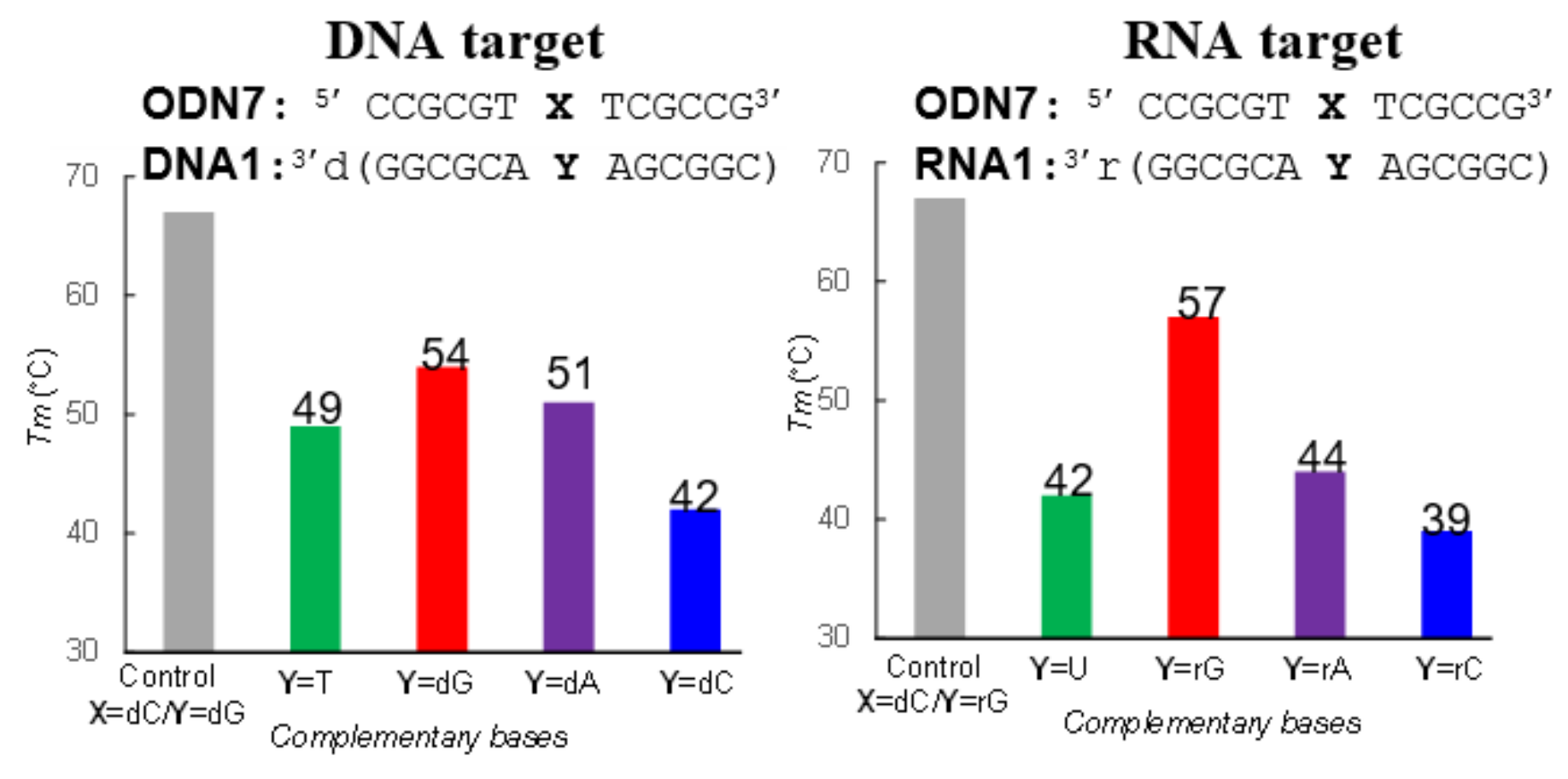
2.5. Tm Measurement
2.6. Alkylation with ODN Probe to HhaI DNMT-1
3. Results
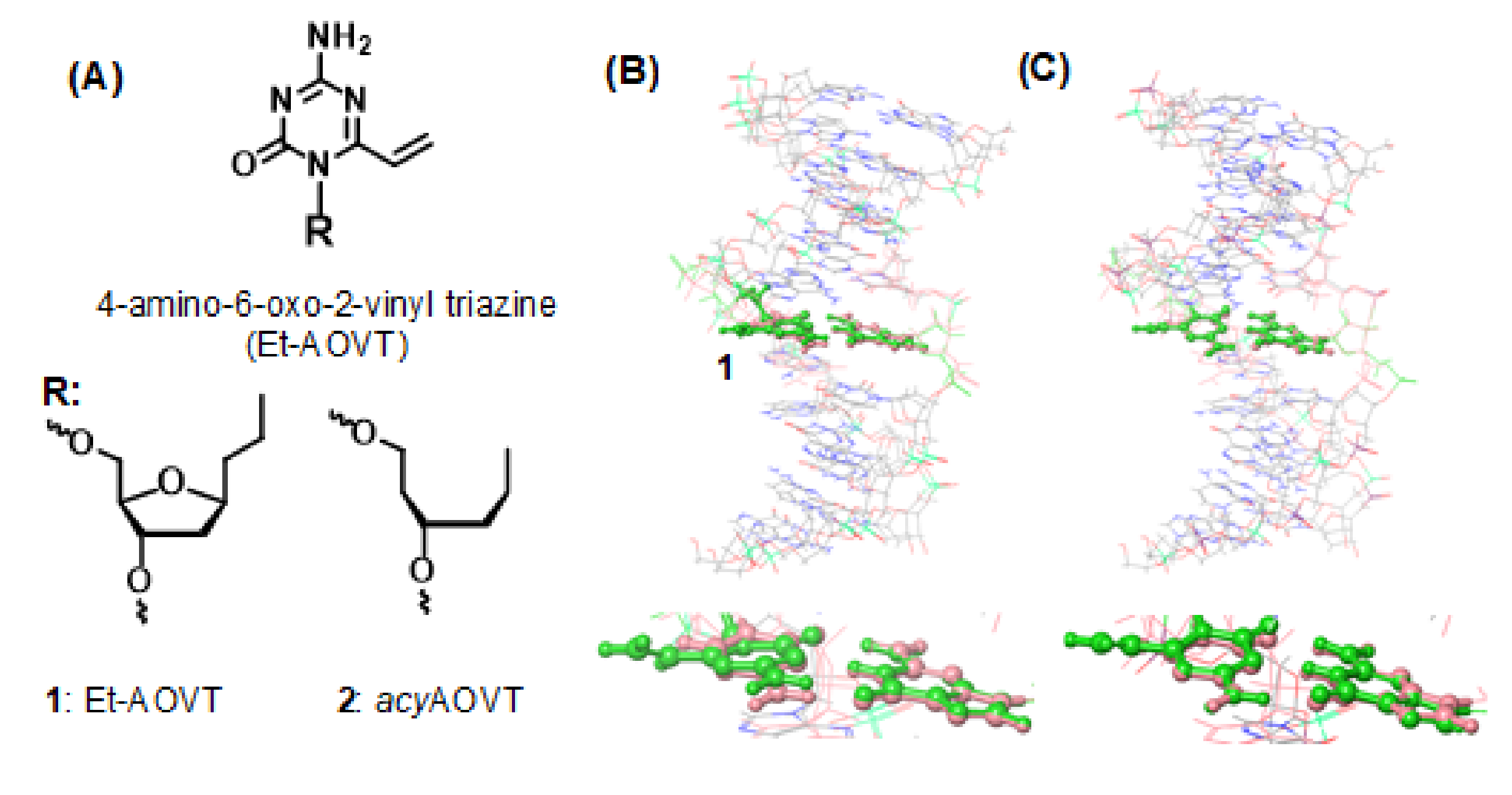
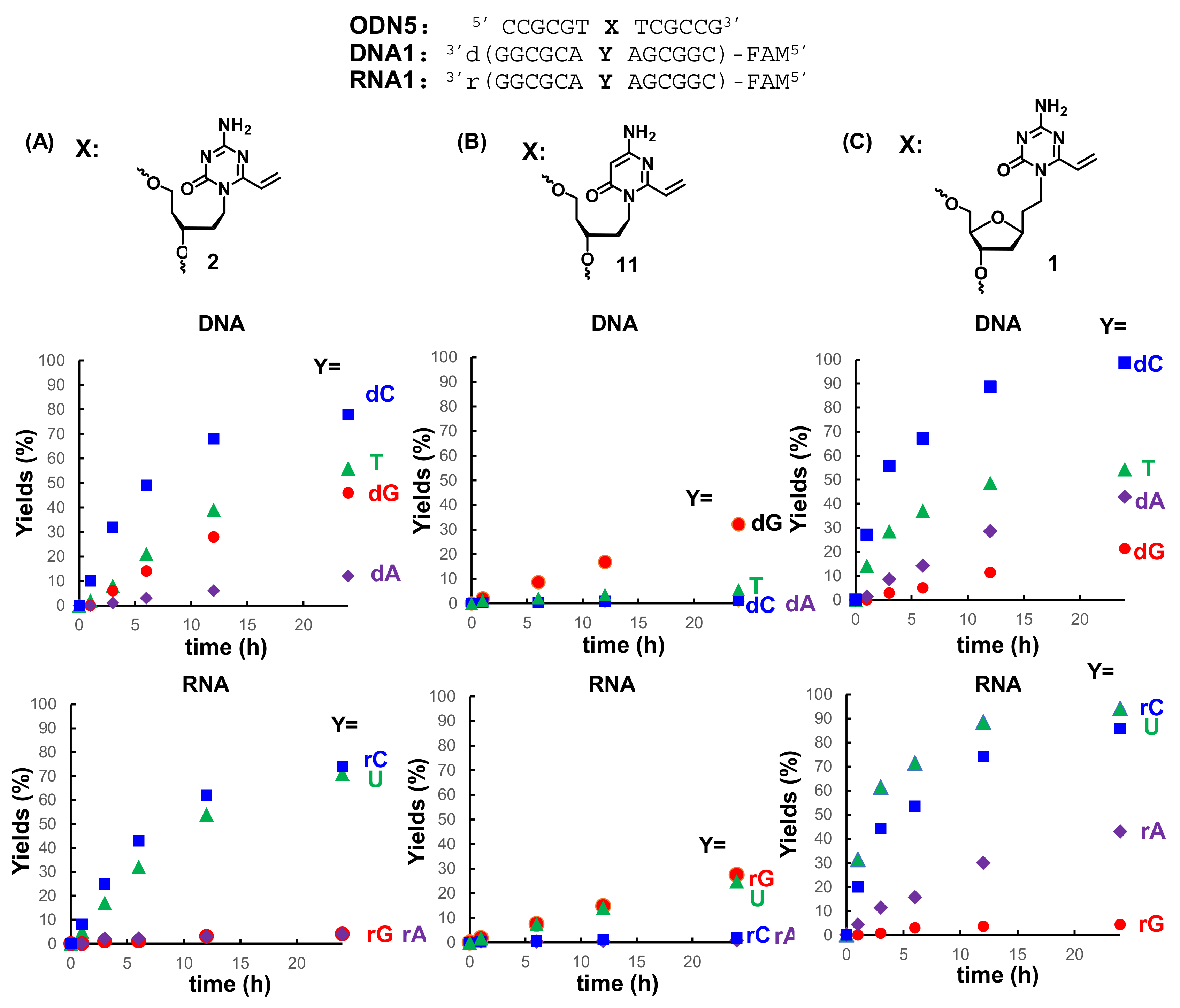
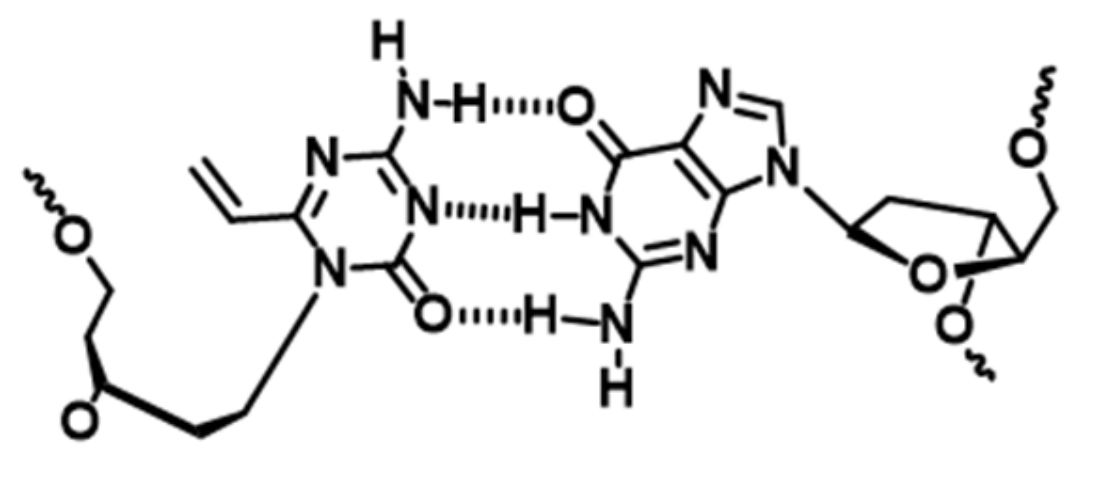
3.1. Synthesis of the acyAOVT Nucleoside Phosphoramidite
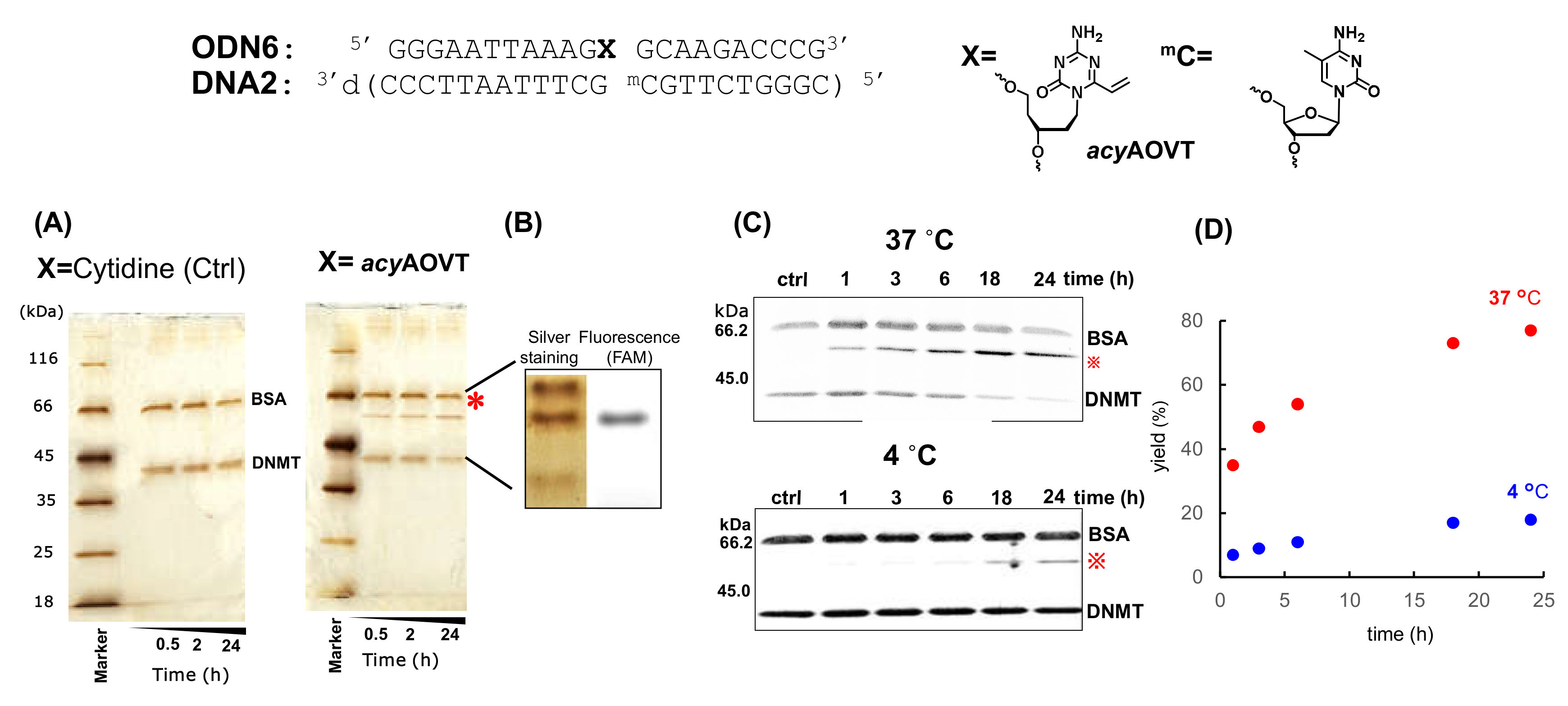
3.2. Synthesis of the Oligonucleotides Containing acyAOVT and Evaluation of the Crosslinking Reactivity
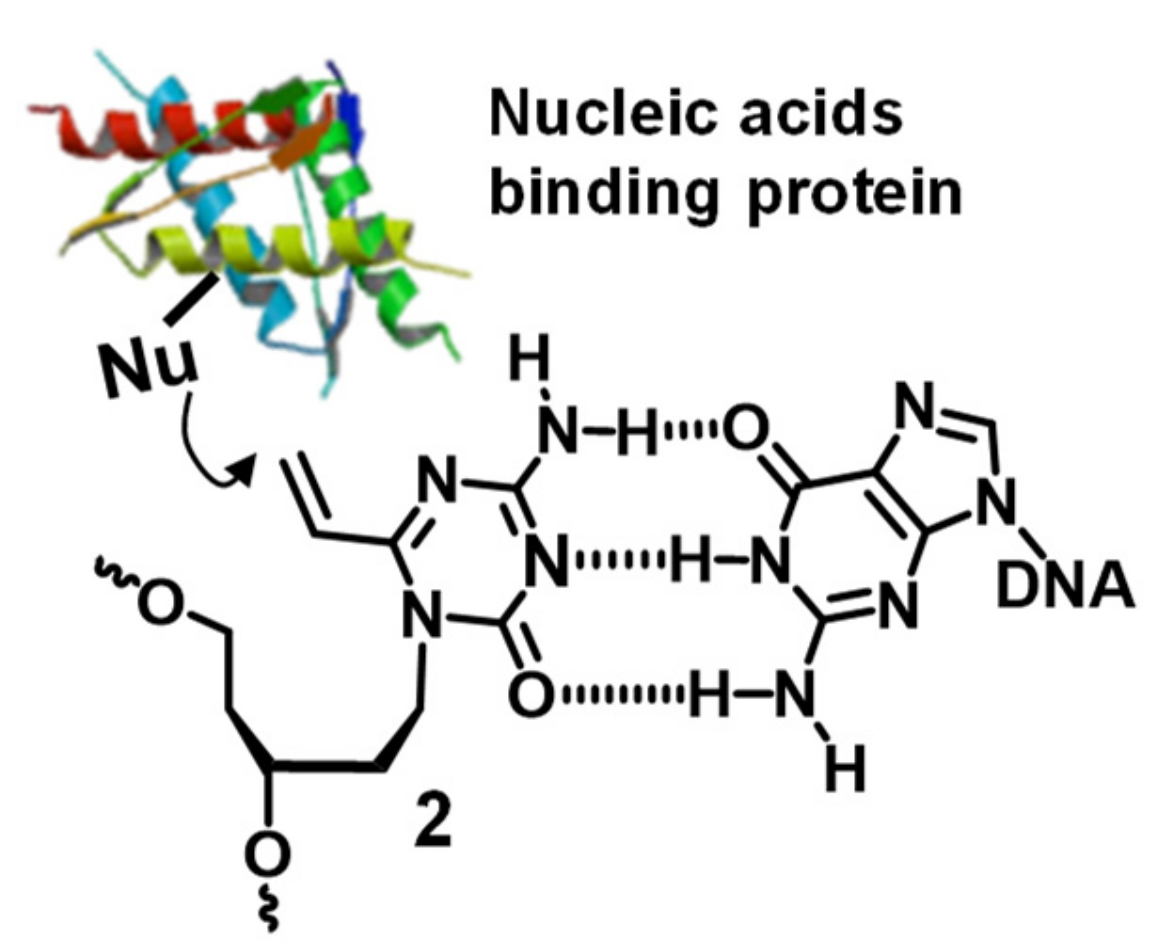
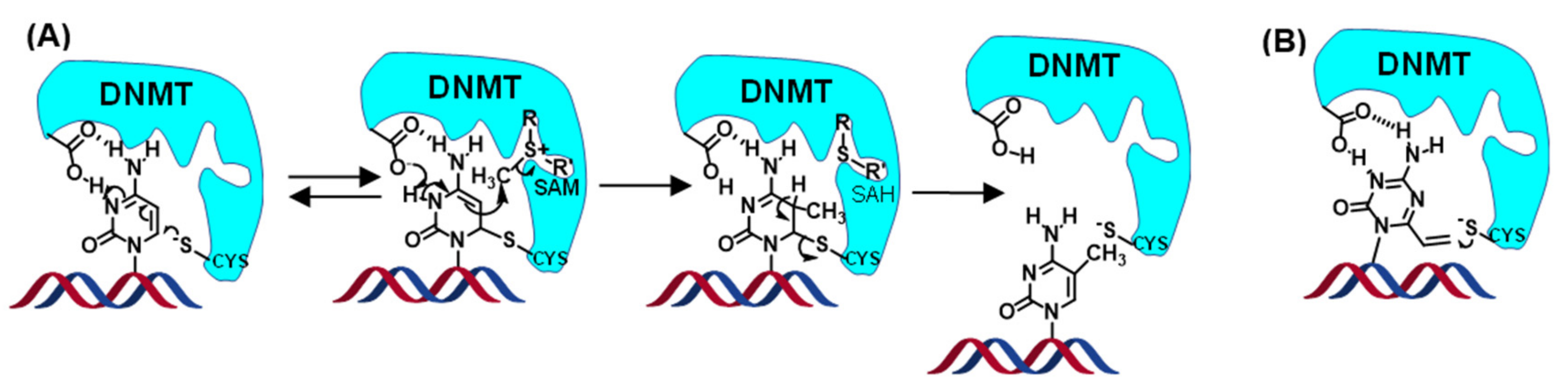
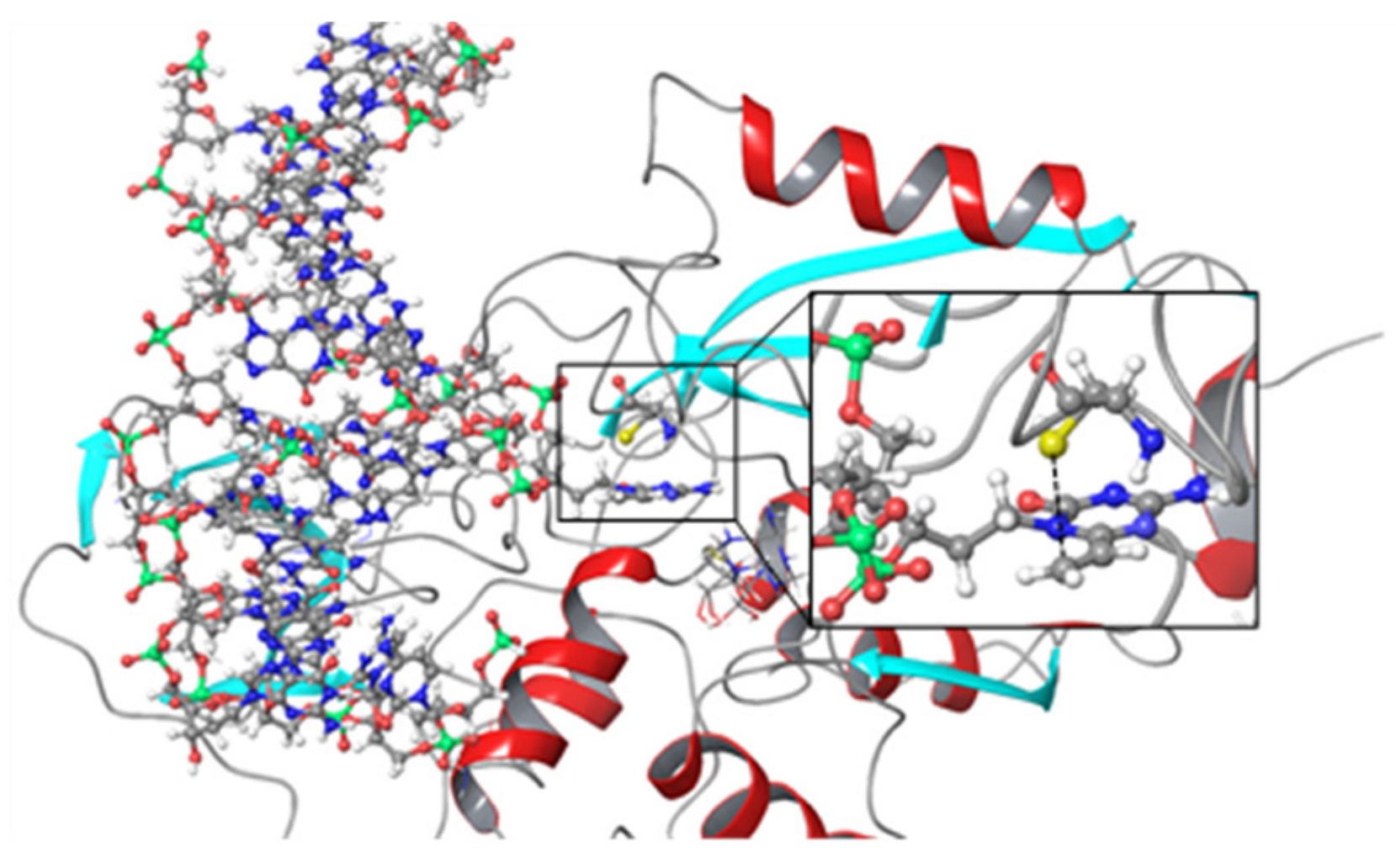
3.3. Crosslinking Reactions to DNA Methyl Transferase
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bouhlel, M.A.; Lambert, M.; David-Cordonnier, M.H. Targeting transcription factor binding to DNA by competing with DNA binders as an approach for controlling gene expression. Curr. Top. Med. Chem. 2015, 15, 1323–1358. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, A.L.; Georges, A.; Veitia, R.A. Transcription factors: Specific DNA binding and specific gene regulation. Trends Genet. 2014, 30, 211–219. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J. Catalytic promiscuity and the divergent evolution of DNA repair enzymes. Chem. Rev. 2006, 106, 720–752. [Google Scholar] [CrossRef] [PubMed]
- Nevinsky, G.A. Structural, thermodynamic, and kinetic basis for the activities of some nucleic acid repair enzymes. J. Mol. Recognit. 2011, 24, 656–677. [Google Scholar] [CrossRef]
- Gujar, H.; Weisenberger, D.J.; Liang, G.N. The roles of human DNA methyltransferases and their isoforms in shaping the epigenome. Genes 2019, 10, 171. [Google Scholar] [CrossRef] [Green Version]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Taylor, K.; Sobczak, K. Intrinsic regulatory role of RNA structural arrangement in alternative splicing control. Int. J. Mol. Sci. 2020, 21, 5161. [Google Scholar] [CrossRef]
- Janakiraman, H.; House, R.P.; Gangaraju, V.K.; Diehl, J.A.; Howe, P.H.; Palanisamy, V. The long (lncRNA) and short (miRNA) of it: TGF beta-mediated control of RNA-binding proteins and noncoding RNAs. Mol. Cancer Res. 2018, 16, 567–579. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.K.; Watanabe, K.; Kitagawa, D. The emerging role of ncRNAs and RNA-binding proteins in Mmitotic apparatus formation. Non Coding RNA 2020, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Sternburg, E.L.; Karginov, F.V. Global approaches in studying RNA-binding protein interaction networks. Trends Biochem. Sci. 2020, 45, 593–603. [Google Scholar] [CrossRef]
- Lercher, L.; McGouran, J.F.; Kessler, B.M.; Schofield, C.J.; Davis, B.G. DNA modification under mild conditions by Suzuki-Miyaura cross-coupling for the generation of functional probes. Angew. Chem. Int. Ed. 2013, 52, 10553–10558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerard-Hirne, T.; Thiebaut, F.; Sachon, E.; Desert, A.; Drujon, T.; Guerineau, V.; Michel, B.Y.; Benhida, R.; Coulon, S.; Saintome, C.; et al. Photoactivatable oligonucleotide probes to trap single-stranded DNA binding proteins: Updating the potential of 4-thiothymidine from a comparative study. Biochimie 2018, 154, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Hollenstein, M.; Leumann, C.J. The synthesis and application of a diazirine-modified uridine analogue for investigating RNA-protein interactions. RSC Adv. 2014, 4, 48228–48235. [Google Scholar] [CrossRef] [Green Version]
- Dziuba, D.; Hoffmann, J.E.; Hentze, M.W.; Schultz, C.A. Genetically Encoded Diazirine Analogue for RNA-Protein Photo-crosslinking. Chembiochem 2020, 21, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Wickramaratne, S.; Mukherjee, S.; Villalta, P.W.; Scharer, O.D.; Tretyakova, N.Y. Synthesis of sequence-specific DNA-protein conjugates via a reductive amination strategy. Bioconj. Chem. 2013, 24, 1496–1506. [Google Scholar] [CrossRef]
- Carrette, L.L.G.; Morii, T.; Madder, A. Toxicity inspired cross-linking for probing DNA-peptide interactions. Bioconj. Chem. 2013, 24, 2008–2014. [Google Scholar] [CrossRef]
- Dadova, J.; Orsag, P.; Pohl, R.; Brazdova, M.; Fojta, M.; Hocek, M. Vinylsulfonamide and acrylamide modification of DNA for cross-linking with proteins. Angew. Chem. Int. Ed. 2013, 52, 10515–10518. [Google Scholar] [CrossRef]
- Olszewska, A.; Pohl, R.; Brazdova, M.; Fojta, M.; Hocek, M. Chloroacetamide-linked nucleotides and DNA for cross-linking with peptides and proteins. Bioconj. Chem. 2016, 27, 2089–2094. [Google Scholar] [CrossRef]
- Matyasovsky, J.; Hocek, M. 2-Substituted 2 ′-deoxyinosine 5 ′-triphosphates as substrates for polymerase synthesis of minor-groove-modified DNA and effects on restriction endonuclease cleavage. Org. Biomol. Chem. 2020, 18, 255–262. [Google Scholar] [CrossRef]
- Yamada, K.; Ishiyama, S.; Onizuka, K.; Nagatsugi, F. Synthesis and properties of cross-linkable DNA duplex using 4-amino-2-oxo-6-vinyl-1,3,5-triazine. Tetrahedron 2017, 73, 1424–1435. [Google Scholar] [CrossRef]
- Kusano, S.; Ishiyama, S.; Lam, S.L.; Mashima, T.; Katahira, M.; Miyamoto, K.; Aida, M.; Nagatsugi, F. Crosslinking reactions of 4-amino-6-oxo-2-vinylpyrimidine with guanine derivatives and structural analysis of the adducts. Nucl. Acids Res. 2015, 43, 7717–7730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, G.P. Defining driver DNA methylation changes in human cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.H.; Li, H.Q.; Liu, F. DNA Methyltransferase inhibitors and their therapeutic potential. Curr. Top. Med. Chem. 2018, 18, 2448–2457. [Google Scholar] [CrossRef] [PubMed]
- Klimasauskas, S.; Kumar Roberts, R.J.; Cheng, X. Hhal methyltransferase flips its target base out of the DNA helix. Cell 1994, 76, 357–369. [Google Scholar] [CrossRef]
- Shigdel, U.K.; He, C. A New 1′-methylenedisulfide deoxyribose that forms an efficient cross-link to DNA cytosine-5-methyltransferase (DNMT). J. Am. Chem. Soc. 2008, 130, 17634–17635. [Google Scholar] [CrossRef]
- Sato, K.; Kawamoto, K.; Shimamura, S.; Ichikawa, S.; Matsuda, A. An oligodeoxyribonucleotide containing 5-formyl-2’-deoxycytidine (fC) at the CpG site forms a covalent complex with DNA cytosine-5 methyltransferases (DNMTs). Bioorg. Med. Chem. Lett. 2016, 26, 5395–5398. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Kunitomo, Y.; Kasai, Y.; Utsumi, S.; Suetake, I.; Tajima, S.; Ichikawa, S.; Matsuda, A. Mechanism-based inhibitor of DNA cytosine-5-methyltransferase by a SNAr reaction with an oligodeoxyribonucleotide containing a 2-amino-4-halopyridine-C-nucleoside. Chembiochem 2018, 19, 865–872. [Google Scholar] [CrossRef]
- Utsumi, S.; Sato, K.; Ichikawa, S. Insight into the recognition mechanism of DNA cytosine-5 methyltransferases (DNMTs) by incorporation of acyclic 5-fluorocytosine (C-F) nucleosides into DNA. Bioorg. Med. Chem. Lett. 2018, 28, 2189–2194. [Google Scholar] [CrossRef]
- Wildenhof, T.M.; Schiffers, S.; Traube, F.R.; Mayer, P.; Carell, T. Influencing epigenetic information with a hydrolytically stable carbocyclic 5-aza-2’-deoxycytidine. Angew. Chem. Int. Ed. 2019, 58, 12984–12987. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odaira, K.; Yamada, K.; Ishiyama, S.; Okamura, H.; Nagatsugi, F. Design of the Crosslinking Reactions for Nucleic Acids-Binding Protein and Evaluation of the Reactivity. Appl. Sci. 2020, 10, 7709. https://doi.org/10.3390/app10217709
Odaira K, Yamada K, Ishiyama S, Okamura H, Nagatsugi F. Design of the Crosslinking Reactions for Nucleic Acids-Binding Protein and Evaluation of the Reactivity. Applied Sciences. 2020; 10(21):7709. https://doi.org/10.3390/app10217709
Chicago/Turabian StyleOdaira, Kenta, Ken Yamada, Shogo Ishiyama, Hidenori Okamura, and Fumi Nagatsugi. 2020. "Design of the Crosslinking Reactions for Nucleic Acids-Binding Protein and Evaluation of the Reactivity" Applied Sciences 10, no. 21: 7709. https://doi.org/10.3390/app10217709