
Flavonoids from Fig (Ficus carica Linn.) Leaves: The Development of a New Extraction Method and Identification by UPLC-QTOF-MS/MS

 ,
, 
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Apparatus
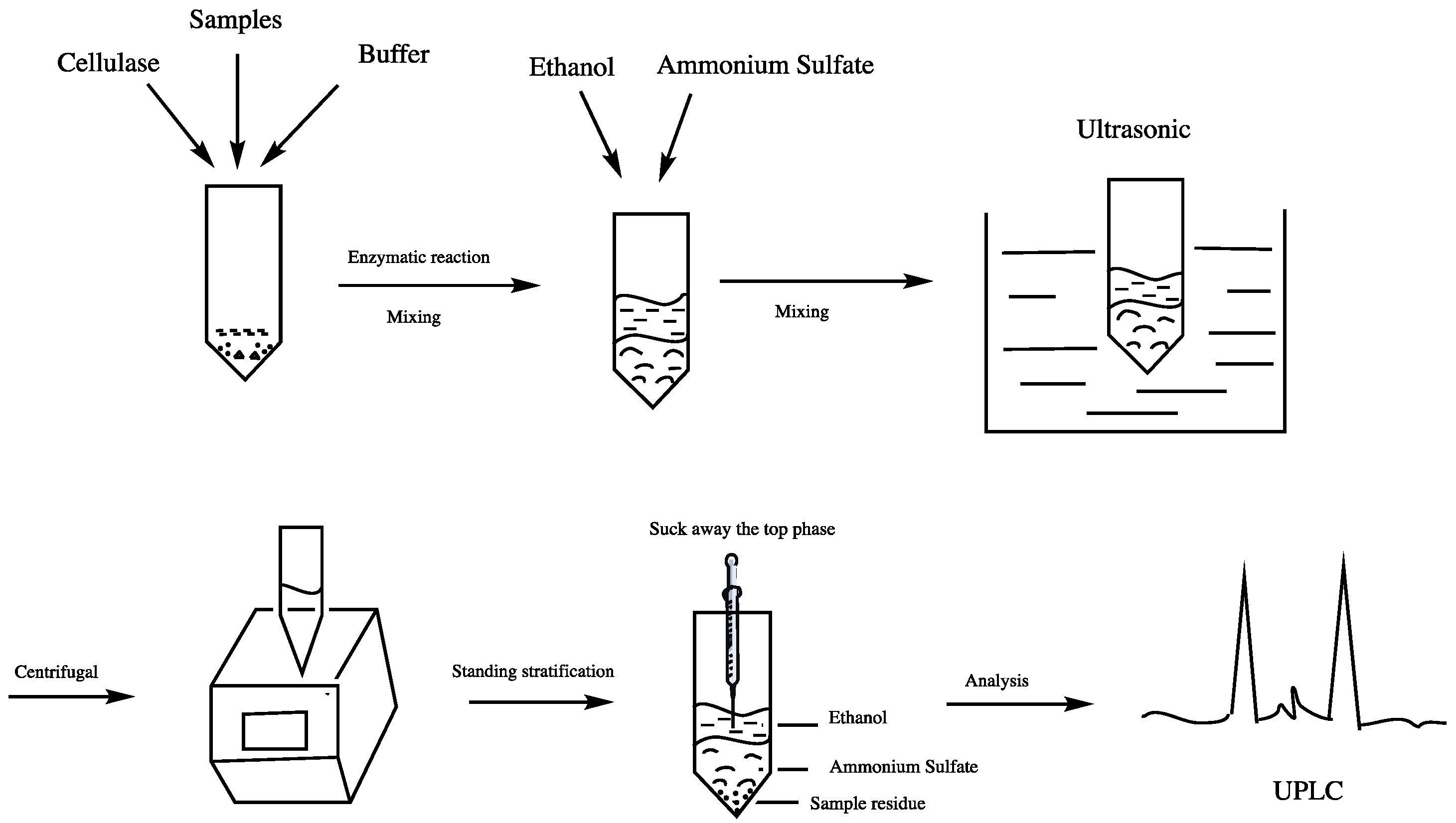
2.3. Ultrasonic Enzyme-Assisted Aqueous Two-Phase Extraction (UEAATPE)
2.4. Determination of Total Flavonoids
2.5. Experiment Design of UEAATPE
2.6. Comparison of Different Extraction Methods
2.7. The Analysis of UPLC-QTOF-MS/MS
2.8. Statistical Analysis
3. Results and Discussion
3.1. Screening of Phase Ratio in ATPS System
3.1.1. Selection of Ethanol Mass Fraction
3.1.2. Selection of Ammonium Sulfate Mass Fraction
3.2. Univariate Analysis of UEAATPE
3.2.1. Effects of Enzyme Concentration on Flavonoid Yield
3.2.2. Effects of Enzymolysis Time on Flavonoid Yield
3.2.3. Effects of Enzymolysis Temperature on Flavonoid Yield
3.2.4. Effects of Ultrasonic Time on Flavonoid Yield
3.2.5. Effects of the Liquid–Solid Ratio on Flavonoid Yield
3.3. Optimization of UEAATPE
3.4. Comparison of Different Methods
3.5. Identification of Flavonoids in Fig Leaves
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Du, C.; Abdullah, J.J.; Greetham, D.; Fu, D.; Yu, M.; Ren, L.; Li, S.; Lu, D. Valorization of food waste into biofertiliser and its field application. J. Clean. Prod. 2018, 187, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Christensen, T.H.; Damgaard, A.; Levis, J.; Zhao, Y.; Bisinella, V. Application of LCA modelling in integrated waste management. Waste Manag. 2020, 118, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Khadivi, A.; Anjam, R.; Anjam, K. Morphological and pomological characterization of edible fig (Ficus carica L.) to select the superior trees. Sci. Hortic. 2018, 238, 66–74. [Google Scholar] [CrossRef]
- Barolo, M.I.; Ruiz Mostacero, N.; López, S.N. Ficus carica L. (Moraceae): An ancient source of food and health. Food Chem. 2014, 164, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.H.; Wu, C.Y.; Cao, Z.Y. Flora of China; Science Press: Beijing, China, 1998; Volume 23, p. 124. [Google Scholar]
- Backes, E.; Leichtweis, M.G.; Pereira, C.; Carocho, M.; Ferreira, I.C.F.R. Ficus carica L. and Prunus spinosa L. extracts as new anthocyanin-based food colorants: A thorough study in confectionery products. Food Chem. 2020, 333, 127457. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, L.; Pereira, C.; Dias, M.I.; Abreu, R.M.V.; Ferreira, I.C.F.R. Nutritional, chemical and bioactive profiles of different parts of a Portuguese common fig (Ficus carica L.) variety. Food Res. Int. 2019, 126, 108572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canal, J.R.; Torres, M.D.; Romero, A.; Pérez, C. A chloroform extract obtained from a decoction of Ficus carica leaves improves the cholesterolaemic status of rats with streptozotocin-induced diabetes. Acta Physi-Ologica Hung. 2000, 87, 71–76. [Google Scholar] [CrossRef]
- Fouad, D.; Alhatem, H.; Abdel-Gaber, R.; Ataya, F. Hepatotoxicity and renal toxicity induced by gamma-radiation and the modulatory protective effect of Ficus carica in male albino rats. Res. Vet. Sci. 2019, 125, 24–35. [Google Scholar] [CrossRef]
- Kais, R.; Dhekra, G.; Dalanda, W.; Slimen, S.; Mohamed, A.; Hichem, S.; Lamjed, M. Ficus carica aqueous extract alleviates delayed gastric emptying and recovers ulcerative colitis-enhanced acute functional gastrointestinal disorders in rats. J. Ethnopharmacol. 2018, 224, 242–249. [Google Scholar]
- Alexandre, E.M.C.; Araújo, P.; Duarte, M.F.; De Freitas, V.; Pintado, M.; Saraiva, J.A. High-pressure assisted extraction of bioactive compounds from industrial fermented fig by-product. J. Food Sci. Technol. 2017, 54, 2519–2531. [Google Scholar] [CrossRef]
- Abbasi, S.; Kamalinejad, M.; Babaie, D.; Shams, S.M.; Sadr, Z.; Gheysari, M.; Askari, V.R.; Rakhshandeh, H. A new topical treatment of atopic dermatitis in pediatric patients based on Ficus carica L. (Fig): A randomized, placebo-controlled clinical trial. Complementary Ther. Med. 2017, 35, 85–91. [Google Scholar] [CrossRef]
- Mustafa, K.; Yu, S.; Zhang, W.; Mohamed, H.; Song, Y. Screening, characterization, and in vitro-ROS dependent cytotoxic potential of extract from Ficus carica against hepatocellular (HepG2) carcinoma cells. S. Afr. J. Bot. 2021, 138, 217–226. [Google Scholar] [CrossRef]
- Patil, V.V.; Patil, V.R. Evaluation of anti-inflammatory activity of Ficus carica Linn. leaves. Indian J. Nat. Prod. Resour. 2011, 2, 151–155. [Google Scholar]
- Pérez, C.; Canal, J.R.; Campillo, J.E.; Romero, A.; Torres, M.D. Hypotriglyceridaemic activity of Ficus carica leaves in experimental hypertriglyceridaemic rats. Phytother. Res. 2015, 13, 188–191. [Google Scholar] [CrossRef]
- Pérez, C.; Domínguez, E.; Ramiro, J.M.; Romero, A.; Campillo, J.E.; Torres, M.D. A study on the glycaemic balance in streptozotocin iabetic rats treated with an aqueous extract of Ficus carica (fig tree) leaves. Phytother. Res. 1996, 10, 82–83. [Google Scholar] [CrossRef]
- Vikas, P. Evaluation of anti-pyretic potential of Ficus carica leaves. Int. J. Pharm. Sci. Rev. Res. 2010, 2, 010. [Google Scholar]
- Caleja, C.; Barros, L.; Prieto, M.A.; Bento, A.; Oliveira, M.B.P.P.; Ferreira, I.C.F.R. Development of a natural preservative obtained from male chestnut flowers: Optimization of a heat-assisted extraction technique. Food Funct. 2019, 10, 1352–1363. [Google Scholar] [CrossRef]
- Soares, R.R.G.; Azevedo, A.M.; Alstine, J.M.V.; Aires-Barros, M.R. Partitioning in aqueous two-phase systems: Analysis of strengths, weaknesses, opportunities and threats. Biotechnol. J. 2015, 10, 1158–1169. [Google Scholar] [CrossRef]
- Liu, Y.; Gong, G.L.; Zhang, J.; Jia, S.Y.; Li, F.; Wang, Y.Y.; Wu, S.H. Response surface optimization of ultrasound-assisted enzymatic extraction polysaccharides from Lycium barbarum. Carbohyd. Polym. 2014, 110, 278–284. [Google Scholar] [CrossRef]
- Chmelová, D.; Škulcová, D.; Legerská, B.; Horník, M.; Ondrejovič, M. Ultrasonic-assisted extraction of polyphenols and antioxidants from Picea abies bark. J. Biotechnol. 2020, 314–315, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Hu, W.-B.; Yang, Z.-W.; Hui, C.; Wang, N.; Liu, X.; Wang, W.-J. Enzymolysis-ultrasonic assisted extraction of flavanoid from Cyclocarya paliurus (Batal) Iljinskaja:HPLC profile, antimicrobial and antioxidant activity. Ind. Crop. Prod. 2019, 130, 615–626. [Google Scholar]
- Chunying, L.; Yukun, Z.; Chunjian, Z.; Yujiao, N.; Kaiting, W.; Jingjing, Z.; Wenyan, Z. Ultrasonic Assisted-Reflux Synergistic Extraction of Camptothecin and Betulinic Acid from Camptotheca acuminata Decne. Fruits. Molecules 2017, 22, 1076. [Google Scholar]
- Luo, X.; Cui, J.; Zhang, H.; Duan, Y.; Zhang, D.; Cai, M.; Chen, G. Ultrasound assisted extraction of polyphenolic compounds from red sorghum ( Sorghum bicolor L.) bran and their biological activities and polyphenolic compositions. Ind. Crop. Prod. 2018, 112, 296–304. [Google Scholar] [CrossRef]
- Orevi, T.; Antov, M. Ultrasound assisted extraction in aqueous two-phase system for the integrated extraction and separation of antioxidants from wheat chaff—ScienceDirect. Sep. Purif. Technol. 2017, 182, 52–58. [Google Scholar]
- Jiao, Y.; Zuo, Y. Ultrasonic extraction and HPLC determination of anthraquinones, aloe-emodine, emodine, rheine, chrysophanol and physcione, in roots of Polygoni multiflori. Phytochem. Anal. 2009, 20, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zuo, Y. Ultrasound-assisted hydrolysis and gas chromatography–mass spectrometric determination of phenolic compounds in cranberry products. Food Chem. 2011, 128, 562–568. [Google Scholar] [CrossRef]
- Song, H.; Yang, R.; Zhao, W.; Katiyo, W.; Hua, X.; Zhang, W. Innovative assistant extraction of flavonoids from pine (Larix olgensis Henry) needles by high-density steam flash-explosion. J. Agric. Food Chem. 2014, 62, 3806. [Google Scholar] [CrossRef]
- Zhang, W.; Zhu, D.; Fan, H.; Liu, X.; Wan, Q.; Wu, X.; Liu, P.; Tang, J.Z. Simultaneous extraction and purification of alkaloids from Sophora flavescens Ait. by microwave-assisted aqueous two-phase extraction with ethanol/ammonia sulfate system. Sep. Purif. Technol. 2015, 141, 113–123. [Google Scholar] [CrossRef]
- Xie, X.; Zhu, D.; Zhang, W.; Huai, W.; Wang, K.; Huang, X.; Zhou, L.; Fan, H. Microwave-assisted aqueous two-phase extraction coupled with high performance liquid chromatography for simultaneous extraction and determination of four flavonoids in Crotalaria sessiliflora L. Ind. Crop. Prod. 2017, 95, 632–642. [Google Scholar] [CrossRef]
- Ma, F.Y.; Gu, C.B.; Li, C.Y.; Luo, M.; Wang, W.; Zu, Y.G.; Li, J.; Fu, Y.J. Microwave-assisted aqueous two-phase extraction of isoflavonoids from Dalbergia odorifera T. Chen leaves. Sep. Purif. Technol. 2013, 115, 136–144. [Google Scholar] [CrossRef]
- Jung, S.; Lamsal, B.P.; Stepien, V.; Johnson, L.A.; Murphy, P.A. Functionality of soy protein produced by enzyme-assisted extraction. J. Am. Oil Chem. Soc. 2006, 83, 71–78. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, Y.; Xu, S.; Xu, W.; Chu, K.; Xu, W.; Zhao, H.; Lu, J. Identification and quantification of phenolic compounds in Vitex negundo L. var. cannabifolia (Siebold et Zucc.) Hand.-Mazz. using liquid chromatography combined with quadrupole time-of-flight and triple quadrupole mass spectrometers. J. Pharm. Biomed. Anal. 2015, 108, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic-Malinovska, R.; Kuzmanova, S.; Winkelhausen, E. Application of ultrasound for enhanced extraction of prebiotic oligosaccharides from selected fruits and vegetables. Ultrason. Sonochem. 2015, 22, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Fabre, N.; Rustan, I.; De, H.E.; Quetin-Leclercq, J. Determination of flavone, flavonol, and flavanone aglycones by negative ion liquid chromatography electrospray ion trap mass spectrometry. J. Am. Soc. Mass Spectrom. 2001, 12, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zuo, Y. Identification of flavonol glycosides in American cranberry fruit. Food Chem. 2007, 101, 1357–1364. [Google Scholar] [CrossRef]
- Zuo, Y.; Lu, X.; Anwar, F.; Hameed, S. Characterization of free and conjugated phenolic compounds in fruits of selected wild plants. Food Chem. 2016, 190, 80–89. [Google Scholar]
- Komatsu, M.; Tomimori, T.; Hatayama, K.; Makiguchi, Y.; Mikuriya, N. Studies on the constituents of Sophora species. II. Constituents of Sophora subprostrata CHUN et T. CHEN. (2). Isolation and structure of new flavonoids, sophoradochromene and sophoranochromene. Chem. Pharm. Bull. 1970, 18, 741–745. [Google Scholar] [CrossRef] [Green Version]
- Adjé, F.A.; Lozano, Y.F.; Gernevé, C.L.; Lozano, P.R.; Meudec, E.; Adima, A.A.; Gaydou, E.M. Phenolic acid and flavonol water extracts of Delonix regia red flowers. Ind. Crop. Prod. 2012, 37, 303–310. [Google Scholar] [CrossRef]
- He, Z.; Xia, W. Analysis of phenolic compounds in chinese olive (Canarium album L.) fruit by RPHPLC-DAD-ESI-MS. Food Chem. 2007, 105, 1307–1311. [Google Scholar] [CrossRef]
- Zhou, C.H.; Luo, Y.Y.; Lei, Z.X.; Wei, G. UHPLC-ESI-MS Analysis of Purified Flavonoids Fraction from Stem of Dendrobium denneaum Paxt. and Its Preliminary Study in Inducing Apoptosis of HepG2 Cells. Evid. -Based Complementary Altern. Med. 2018, 2018, 8936307. [Google Scholar] [CrossRef] [Green Version]
- Sousa, A.D.; Maia, A.I.V.; Rodrigues, T.H.S.; Canuto, K.M.; Ribeiro, P.R.V.; de Cassia Alves Pereira, R.; Vieira, R.F.; de Brito, E.S. Ultrasound-assisted and pressurized liquid extraction of phenolic compounds from Phyllanthus amarus and its composition evaluation by UPLC-QTOF. Ind. Crop. Prod. 2016, 79, 91–103. [Google Scholar] [CrossRef]
- He, L.; Zhang, Z.; Lu, L.; Liu, Y.; Li, S.; Wang, J.; Song, Z.; Yan, Z.; Miao, J. Rapid identification and quantitative analysis of the chemical constituents in Scutellaria indica L. by UHPLC-QTOF-MS and UHPLC-MS/MS. J. Pharm. Biomed. Anal. 2016, 117, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Batista, M.T.; Gomes, E.T. C-glycosylflavones from Ceratonia siliqua cotyledons. Phytochemistry 1993, 34, 1191–1193. [Google Scholar] [CrossRef]
- Norbaek, R.; Brandt, K.; Kondo, T. Identification of flavone C-glycosides including a new flavonoid chromophore from barley leaves (Hordeum vulgare L.) by improved NMR techniques. J. Agric. Food Chem. 2000, 48, 1703–1707. [Google Scholar] [CrossRef] [PubMed]
- Muth, D.; Marsden-Edwards, E.; Kachlicki, P.; Stobiecki, M. Differentiation of isomeric malonylated flavonoid glyconjugates in plant extracts with UPLC-ESI/MS/MS. Phytochem. Anal. 2010, 19, 444–452. [Google Scholar] [CrossRef]
- Huang, D.; Zhou, X.; Si, J.; Gong, X.; Wang, S. Studies on cellulase-ultrasonic assisted extraction technology for flavonoids from Illicium verum residues. Chem. Cent. J. 2016, 10, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Calibration Curve | R2 | Linear Range (μg/mL) | LOD (μg/mL) | LOQ (μg/mL) |
|---|---|---|---|---|---|
| rutin | y = 5.5778 x + 0.0635 | 0.9929 | 3.901–250 | 2.196 | 7.032 |
| No. | Mass Fraction of Ethanol (%) | Mass Fraction of Ammonium Sulfate (%) | Concentration of Enzymes (U/g) | Enzymolysis Time (min) | Enzymolysis Temperature (°C) | Ultrasonic Time (min) | Liquid–Solid Ratio (mL/g) | Yield of Total Flavonoids (mg/g) |
|---|---|---|---|---|---|---|---|---|
| 1 | 32 | 18 | 0.3 | 180 | 50 | 30 | 20 | 56.83 |
| 2 | 34 | 18 | 0.3 | 180 | 50 | 30 | 20 | 58.92 |
| 3 | 36 | 18 | 0.3 | 180 | 50 | 30 | 20 | 59.46 |
| 4 | 38 | 18 | 0.3 | 180 | 50 | 30 | 20 | 60.02 |
| 5 | 40 | 18 | 0.3 | 180 | 50 | 30 | 20 | 58.18 |
| 6 | 38 | 14 | 0.3 | 180 | 50 | 30 | 20 | 47.79 |
| 7 | 38 | 16 | 0.3 | 180 | 50 | 30 | 20 | 57.03 |
| 8 | 38 | 18 | 0.3 | 180 | 50 | 30 | 20 | 60.19 |
| 9 | 38 | 20 | 0.3 | 180 | 50 | 30 | 20 | 58.41 |
| 10 | 38 | 22 | 0.3 | 180 | 50 | 30 | 20 | 55.40 |
| 11 | 38 | 18 | 0.3 | 180 | 50 | 30 | 20 | 47.13 |
| 12 | 38 | 18 | 0.4 | 180 | 50 | 30 | 20 | 60.00 |
| 13 | 38 | 18 | 0.5 | 180 | 50 | 30 | 20 | 59.03 |
| 14 | 38 | 18 | 0.6 | 180 | 50 | 30 | 20 | 57.25 |
| 15 | 38 | 18 | 0.7 | 180 | 50 | 30 | 20 | 57.17 |
| 16 | 38 | 18 | 0.3 | 90 | 50 | 30 | 20 | 46.13 |
| 17 | 38 | 18 | 0.3 | 120 | 50 | 30 | 20 | 50.24 |
| 18 | 38 | 18 | 0.3 | 150 | 50 | 30 | 20 | 53.20 |
| 19 | 38 | 18 | 0.3 | 180 | 50 | 30 | 20 | 59.89 |
| 20 | 38 | 18 | 0.3 | 210 | 50 | 30 | 20 | 50.00 |
| 21 | 38 | 18 | 0.3 | 180 | 35 | 30 | 20 | 41.55 |
| 22 | 38 | 18 | 0.3 | 180 | 40 | 30 | 20 | 44.99 |
| 23 | 38 | 18 | 0.3 | 180 | 45 | 30 | 20 | 58.09 |
| 24 | 38 | 18 | 0.3 | 180 | 50 | 30 | 20 | 60.12 |
| 25 | 38 | 18 | 0.3 | 180 | 55 | 30 | 20 | 52.53 |
| 26 | 38 | 18 | 0.3 | 180 | 50 | 10 | 20 | 56.11 |
| 27 | 38 | 18 | 0.3 | 180 | 50 | 20 | 20 | 57.99 |
| 28 | 38 | 18 | 0.3 | 180 | 50 | 30 | 20 | 60.91 |
| 29 | 38 | 18 | 0.3 | 180 | 50 | 40 | 20 | 59.23 |
| 30 | 38 | 18 | 0.3 | 180 | 50 | 50 | 20 | 57.10 |
| 31 | 38 | 18 | 0.3 | 180 | 50 | 30 | 10 | 55.01 |
| 32 | 38 | 18 | 0.3 | 180 | 50 | 30 | 20 | 60.01 |
| 33 | 38 | 18 | 0.3 | 180 | 50 | 30 | 30 | 58.32 |
| 34 | 38 | 18 | 0.3 | 180 | 50 | 30 | 40 | 58.29 |
| 35 | 38 | 18 | 0.3 | 180 | 50 | 30 | 50 | 58.10 |
| Runs | Independent Variables | Yield of Total Flavonoids (mg/g) | |||
|---|---|---|---|---|---|
| Ethanol Concentration (X1, %) | Ammonium Sulfate Concentration (X2, %) | Liquid—Solid Ratio (X3, mL/g) | Ultrasonic Time (X4, min) | ||
| 1 | 38 | 18 | 30:1 | 20 | 43.71 |
| 2 | 38 | 18 | 10:1 | 20 | 44.62 |
| 3 | 36 | 18 | 10:1 | 30 | 52.80 |
| 4 | 38 | 16 | 20:1 | 40 | 45.44 |
| 5 | 36 | 18 | 30:1 | 30 | 35.25 |
| 6 | 38 | 16 | 10:1 | 30 | 49.99 |
| 7 | 40 | 18 | 30:1 | 30 | 42.99 |
| 8 | 40 | 18 | 10:1 | 30 | 48.75 |
| 9 | 38 | 20 | 10:1 | 30 | 48.13 |
| 10 | 36 | 18 | 20:1 | 20 | 48.74 |
| 11 | 38 | 18 | 20:1 | 30 | 60.39 |
| 12 | 40 | 18 | 20:1 | 20 | 42.87 |
| 13 | 40 | 18 | 20:1 | 40 | 48.16 |
| 14 | 38 | 18 | 10:1 | 40 | 49.70 |
| 15 | 38 | 20 | 20:1 | 40 | 45.84 |
| 16 | 38 | 16 | 30:1 | 30 | 39.54 |
| 17 | 38 | 20 | 30:1 | 30 | 45.68 |
| 18 | 38 | 18 | 30:1 | 40 | 30.87 |
| 19 | 36 | 18 | 20:1 | 40 | 41.04 |
| 20 | 40 | 16 | 20:1 | 30 | 49.59 |
| 21 | 38 | 18 | 20:1 | 30 | 58.94 |
| 22 | 38 | 18 | 20:1 | 30 | 59.11 |
| 23 | 36 | 20 | 20:1 | 30 | 51.40 |
| 24 | 40 | 20 | 20:1 | 30 | 51.11 |
| 25 | 38 | 16 | 20:1 | 20 | 48.90 |
| 26 | 38 | 18 | 20:1 | 30 | 61.08 |
| 27 | 36 | 16 | 20:1 | 30 | 54.60 |
| 28 | 38 | 18 | 20:1 | 30 | 58.50 |
| 29 | 38 | 20 | 20:1 | 20 | 51.70 |
| Source | Sum of Squares | Mean Square | F-Value | p-Value | Significance |
|---|---|---|---|---|---|
| Model | 1.40 × 103 | 100.32 | 28.36 | <0.0001 | significant |
| X1 | 1.05 × 10−2 | 1.05 × 10−2 | 2.97 × 10−3 | 0.9573 | not significant |
| X2 | 2.81 | 2.81 | 0.79 | 0.3882 | not significant |
| X3 | 260.89 | 260.89 | 73.76 | <0.0001 | significant |
| X4 | 31.69 | 31.69 | 8.96 | 0.0097 | not significant |
| X1 X2 | 5.57 | 5.57 | 1.58 | 0.2301 | not significant |
| X1 X3 | 34.76 | 34.76 | 9.83 | 0.0073 | not significant |
| X1 X4 | 42.11 | 42.11 | 11.91 | 0.0039 | not significant |
| X2 X3 | 16.00 | 16.00 | 4.52 | 0.0517 | not significant |
| X2 X4 | 1.44 | 1.44 | 0.41 | 0.5342 | not significant |
| X3 X4 | 80.28 | 80.28 | 22.70 | 0.0003 | not significant |
| X12 | 175.31 | 1.75 × 102 | 49.57 | <0.0001 | significant |
| X22 | 73.68 | 73.68 | 20.83 | 0.0004 | not significant |
| X32 | 598.54 | 5.99 × 102 | 1.70 × 102 | <0.0001 | significant |
| X42 | 459.01 | 4.60 × 102 | 1.30 × 102 | <0.0001 | significant |
| Lack of Fit | 44.82 | 4.48 | 3.82 | 0.1042 | not significant |
| R2 | 0.9659 | ||||
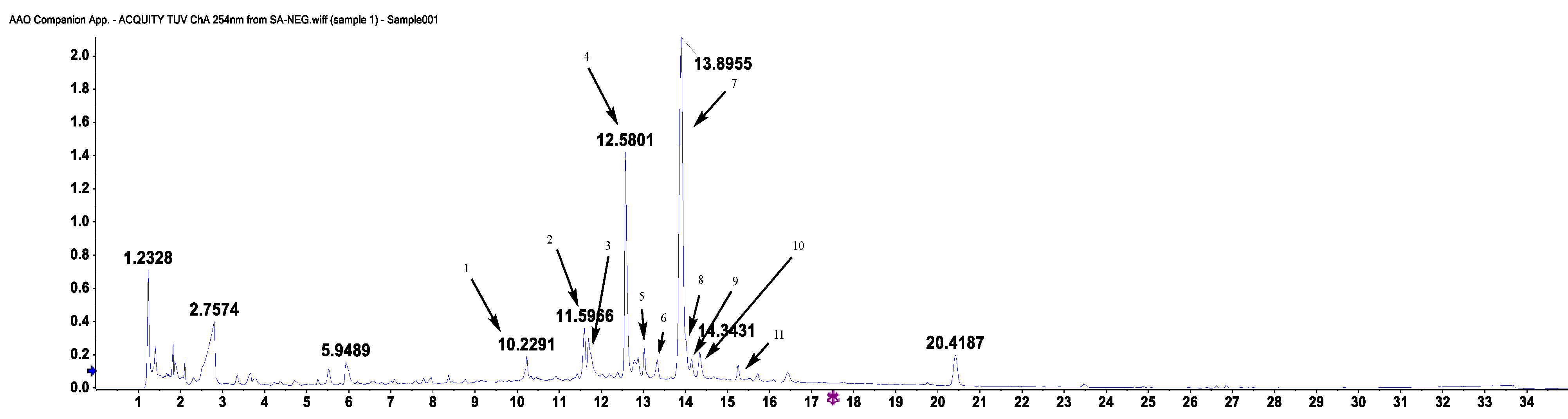
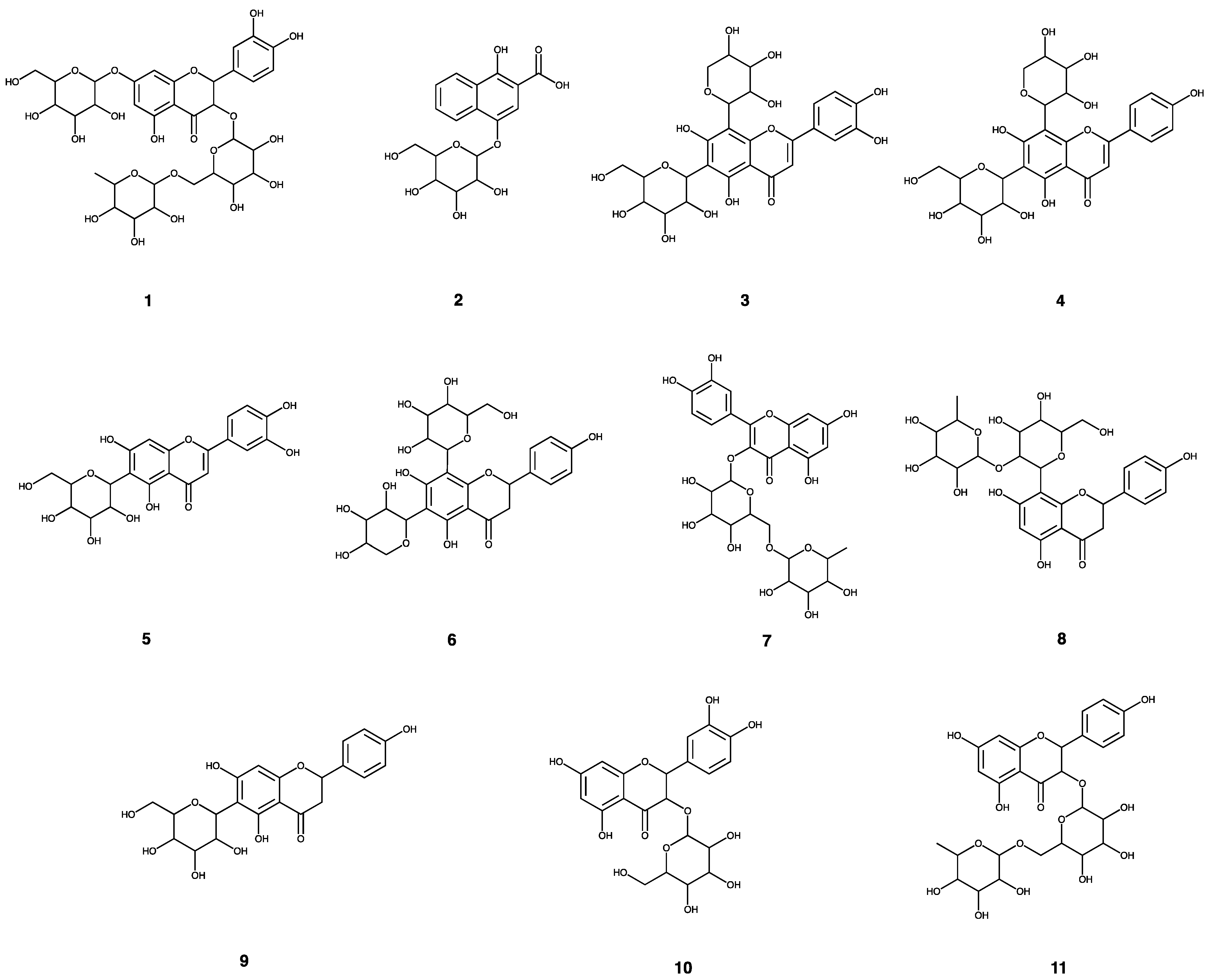
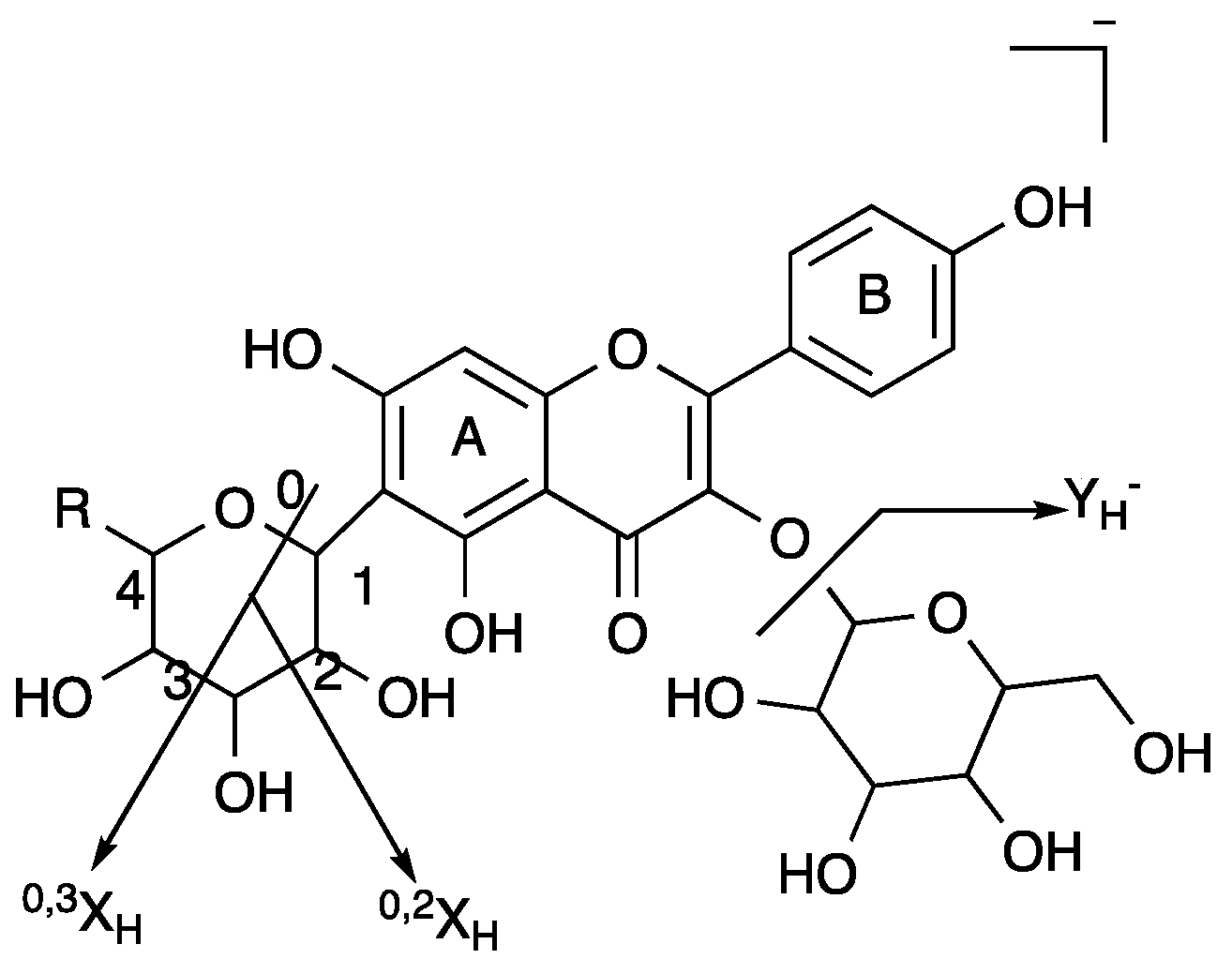
| Peak No. | tR | MS | MS/MS | Molecular Weight | Molecular Formula | Identification |
|---|---|---|---|---|---|---|
| 1 | 10.22 | 771.2027 | 609.1530, 462.0838, 301.0357 | 772.20621 | C33H40O21 | 3-O-(rhamnopyranosyl-glucopyranosyl)-7-O-(glucopyrnosyl)-quercetin |
| 2 | 11.59 | 365.0881 | 203.0352, 159.0454, 130.0422 | 366.09508 | C17H18O9 | 2-carboxyl-1,4-naphthohydroquinone-4-O-hexoside |
| 3 | 11.7 | 579.1366 | 519.1194, 489.1083, 429.0856, 369.0635 | 580.14282 | C26H28O15 | luteolin 6-C-hexoside, 8-C-pentoside |
| 4 | 12.58 | 563.1414 | 473.1115,443.1001, 353.0670 | 564.14791 | C26H28O14 | kaempferol 6-C-hexoside-8-C-hexoside |
| 5 | 12.87 | 447.0934 | 369.0615, 357.0622, 327.0511, 297.0397, 285.0396, 133.0280 | 448.10050 | C21H20O11 | quercetin 6-C-hexobioside |
| 6 | 13.02 | 563.1412 | 443.1001, 353.0670 | 564.14791 | C26H28O14 | kaempferol 6-C-hexoside-8-C-hexoside |
| 7 | 13.89 | 609.1470 | 301.0362, 151.0031, 257.0450, 273.0477 | 610.15338 | C27H30O16 | quercetin 3-O-hexobioside |
| 8 | 14.02 | 577.1577 | 457.1164, 293.0454 | 578.16356 | C27H30O14 | apigenin 2″-O-pentoside |
| 9 | 14.14 | 432.1056 | 341.0673, 311.0564, 283.0612 | 432.10565 | C21H20O10 | apigenin 6-C-hexoside |
| 10 | 14.34 | 463.0890 | 301.0357 | 464.09548 | C21H20O12 | quercetin 3-O-hexoside |
| 11 | 15.25 | 593.1524 | 285.0403 | 594.15847 | C27H30O15 | kaempferol 3-O-hexobioside |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, C.; Li, S.; Li, C.; Wang, T.; Tian, Y.; Li, X. Flavonoids from Fig (Ficus carica Linn.) Leaves: The Development of a New Extraction Method and Identification by UPLC-QTOF-MS/MS. Appl. Sci. 2021, 11, 7718. https://doi.org/10.3390/app11167718
Zhao C, Li S, Li C, Wang T, Tian Y, Li X. Flavonoids from Fig (Ficus carica Linn.) Leaves: The Development of a New Extraction Method and Identification by UPLC-QTOF-MS/MS. Applied Sciences. 2021; 11(16):7718. https://doi.org/10.3390/app11167718
Chicago/Turabian StyleZhao, Chunjian, Shen Li, Chunying Li, Tingting Wang, Yao Tian, and Xin Li. 2021. "Flavonoids from Fig (Ficus carica Linn.) Leaves: The Development of a New Extraction Method and Identification by UPLC-QTOF-MS/MS" Applied Sciences 11, no. 16: 7718. https://doi.org/10.3390/app11167718
APA StyleZhao, C., Li, S., Li, C., Wang, T., Tian, Y., & Li, X. (2021). Flavonoids from Fig (Ficus carica Linn.) Leaves: The Development of a New Extraction Method and Identification by UPLC-QTOF-MS/MS. Applied Sciences, 11(16), 7718. https://doi.org/10.3390/app11167718