
Monitoring Scheme for the Detection of Hydrogen Leakage from a Deep Underground Storage. Part 2: Physico-Chemical Impacts of Hydrogen Injection into a Shallow Chalky Aquifer

 , , ,
, , ,
Abstract
Featured Application
Abstract
1. Introduction
1.1. General Information on Underground Hydrogen Storage
1.2. Hydrogen Reactivity in a Natural Environment
1.3. Risk of Contamination of Drinking Water Aquifers
- Reduction of nitrates (NO3−) to nitrites (NO2−), or even to ammonium (NH4+), and then to gaseous nitrogen (N2)
- Reduction of sulfates (SO42−) to sulfides (SO3−), or even to hydrogen sulfide (H2S)
- Reduction of iron III to iron II
- Dissolution of metallic trace elements potentially present in the aquifer rock following the drop in the redox potential
2. Material and Methods
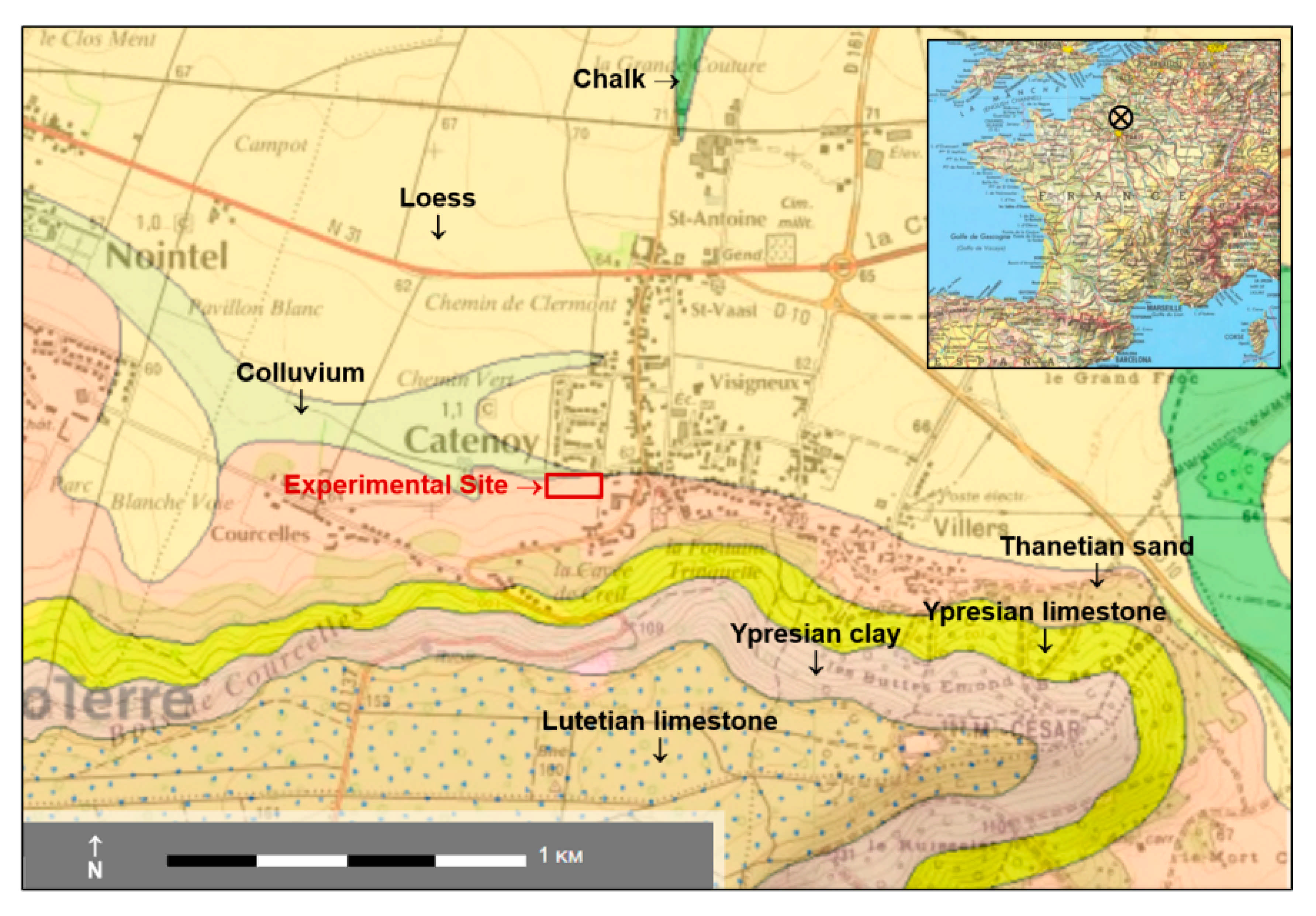
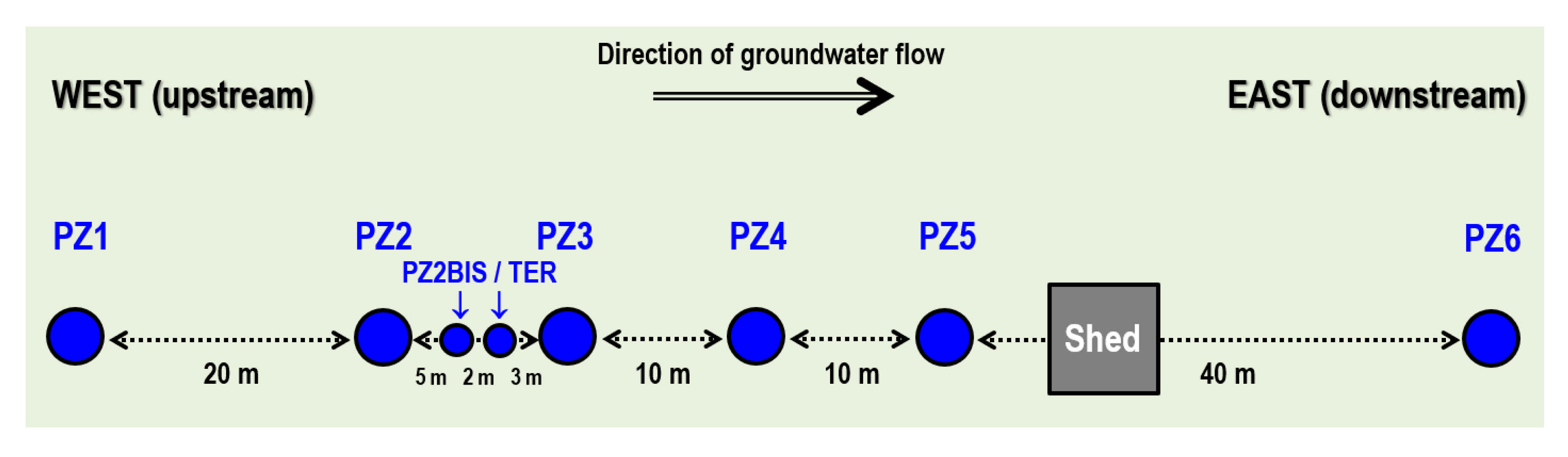
2.1. Overview of the Site Context and Equipment
2.2. Overview of the Monitoring Protocol
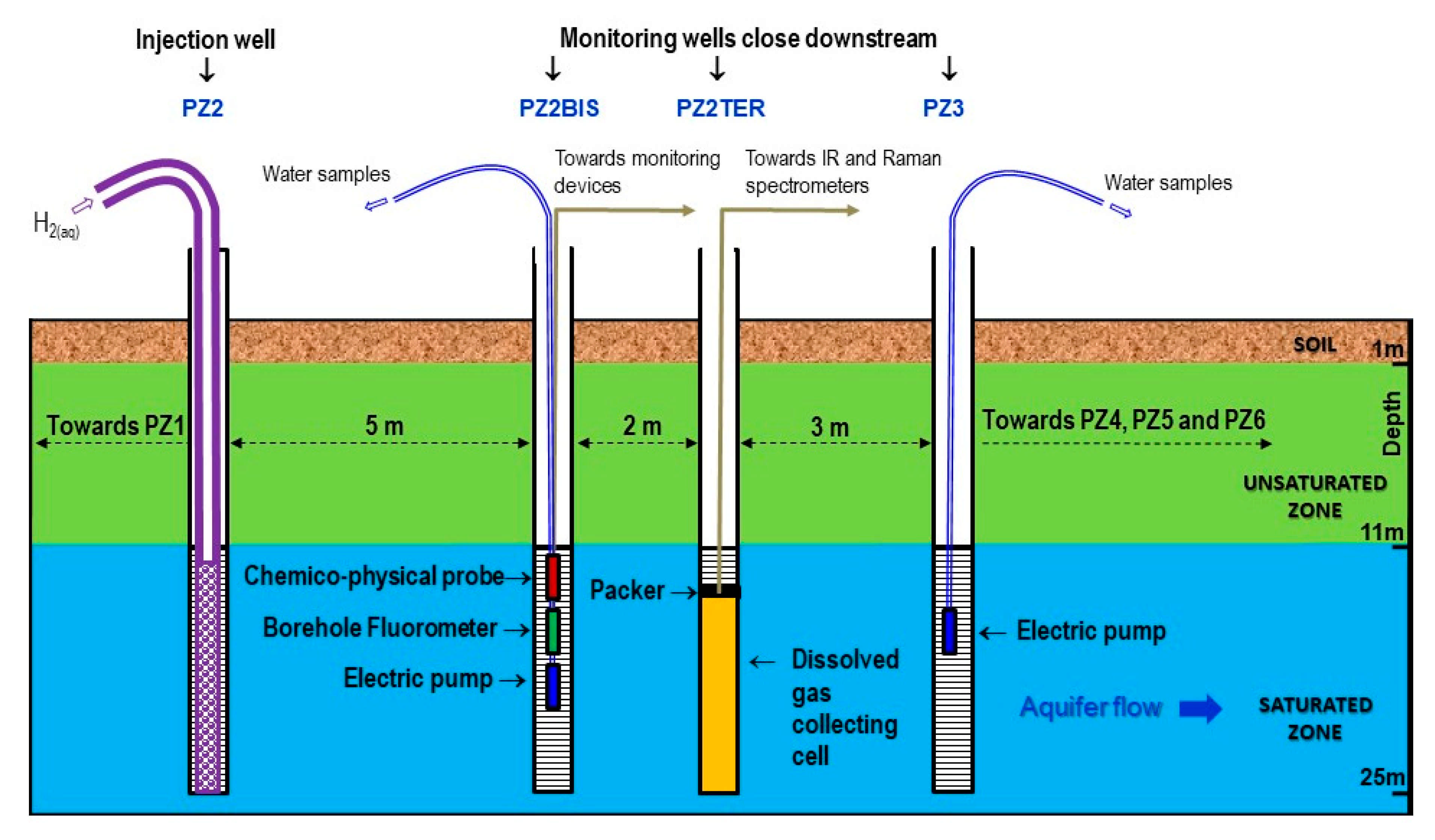
- Two physicochemical probes measure temperature, pH, electrical conductivity, oxidation-reduction potential, and dissolved oxygen. One is installed in the PZ2bis while the other is mobile in order to take measurements at all the piezometers. It should be noted that this last probe had to be replaced on 19 November 2019 at 14:00, which is visible on the graphs as a deviation due to the new calibration (just before the injection of dissolved hydrogen).
- Two field fluorometers allow on site measure of the water fluorescence. As before, one is installed in the PZ2bis, while the other is mobile in order to take measurements in all the piezometers by groundwater sampling.
- Six submerged electric pumps are installed at a depth of 16 m at the PZ1, PZ2BIS, PZ3, PZ4, PZ5 and PZ6 piezometers. They are used to regularly sample the groundwater in order to perform laboratory analyses of tracers, dissolved gases (CH4, He, H2, and H2S), major elements (Ca2+, Mg2+, Na+, K+, HCO3−, Cl−, SO42−, and NO3−, with detection limits of 0.01–0.05 mg·L−1), and minor elements (SO3−, S2−, NO2−, and NH4+, with detection limits of 0.01–0.02 mg·L−1).
- Raman and Infrared (IR) spectrometers installed at the PZ2TER [25,27] make it possible to analyze the concentration in the groundwater of mononuclear diatomic molecules (H2, O2, and N2 for Raman) and polar molecules (CO2 and CH4 for Raman and IR). It should be noted that the data acquired by these devices are not presented here but are the subject of another specific publication to come. Here, we selected Raman and IR technologies because they were already used in other studies, which ensures they suit our needs. New sensing technologies using optical fiber grating platforms are presently developed and tested on lab benches and would be promising for field studies in the close future [28].
- There are specific analyzers for measuring the gas concentration in the piezometer head spaces and in the gas mixture released from the water extracted from the aquifer. A DRÄGER multigas analyzer equipped with a catalytic cell (resolution of 0.1% vol.) and a portable Biogas analyzer equipped with an electrochemical cell (resolution of 1 ppm) are used for hydrogen, while an ALCATEL ASM 122D transportable mass spectrometer is used for helium.
- Device for extracting and degassing water by mechanical agitation serves to establish the dissolved gas concentration in the groundwater, in conjunction with the gas analyzers detailed above.
2.3. Experimental Protocol
3. Results
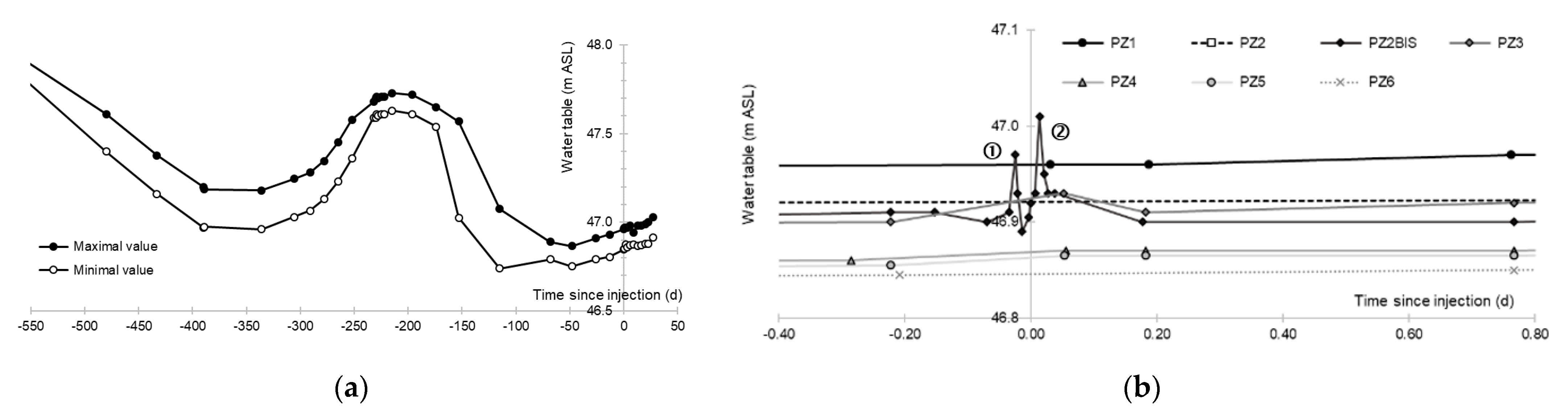
3.1. Aquifer Piezometry
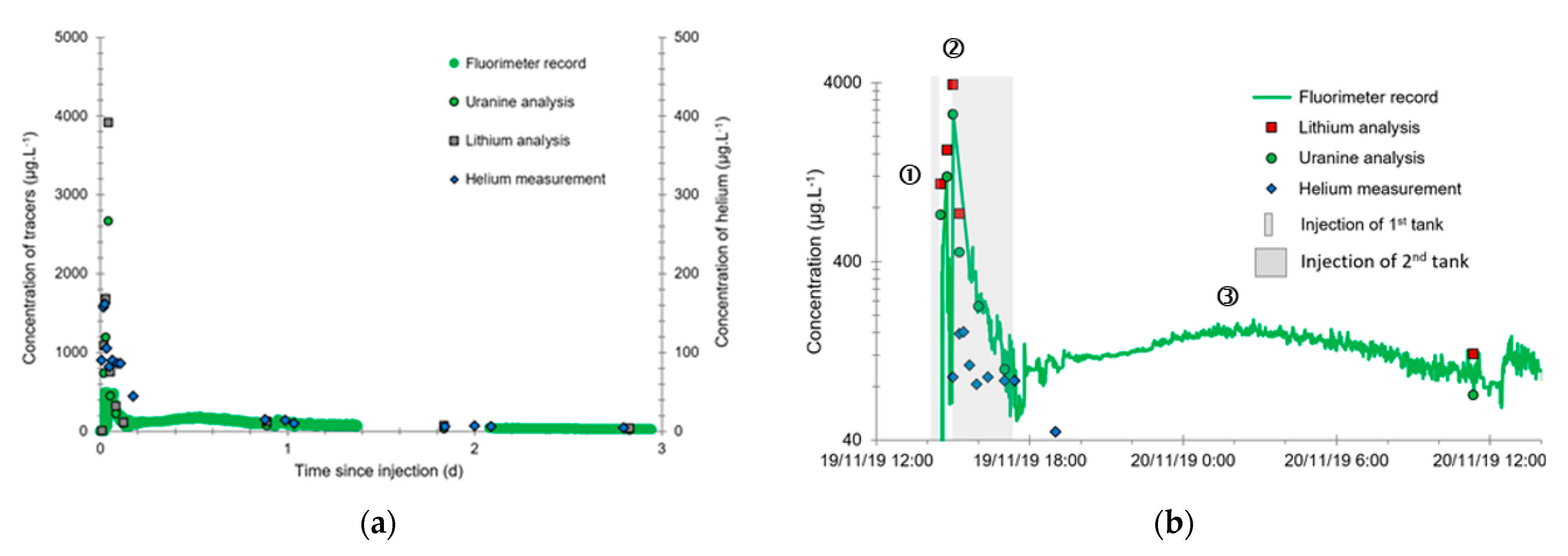
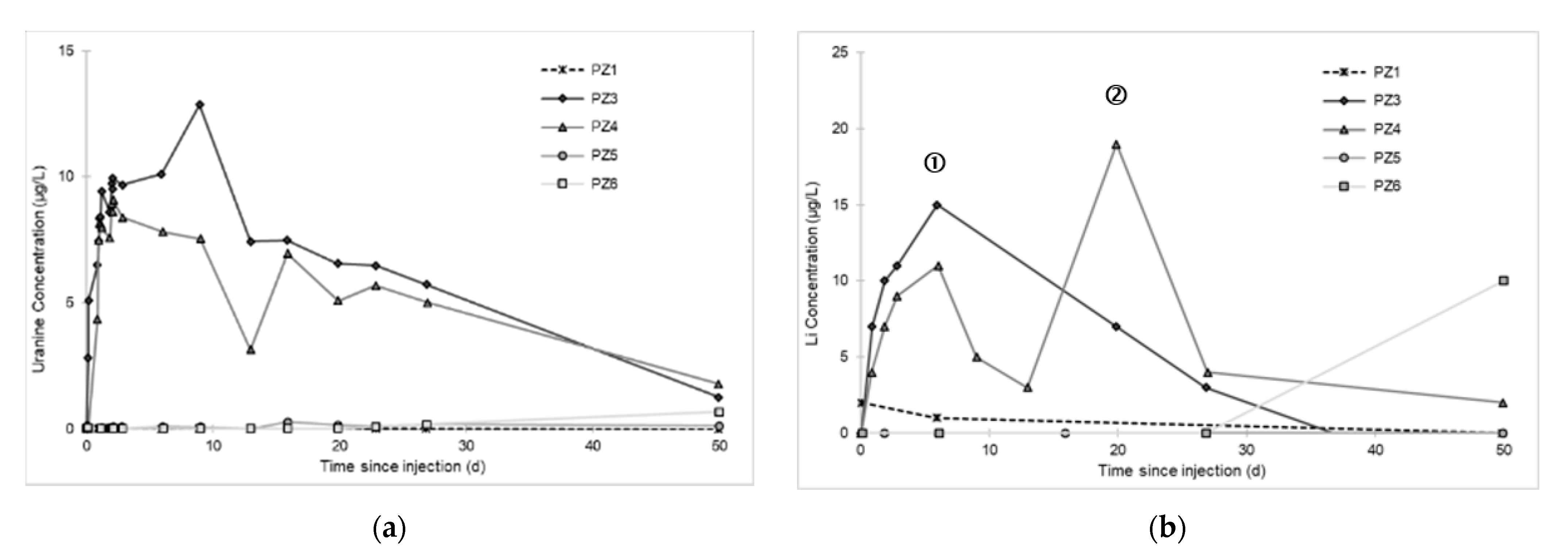
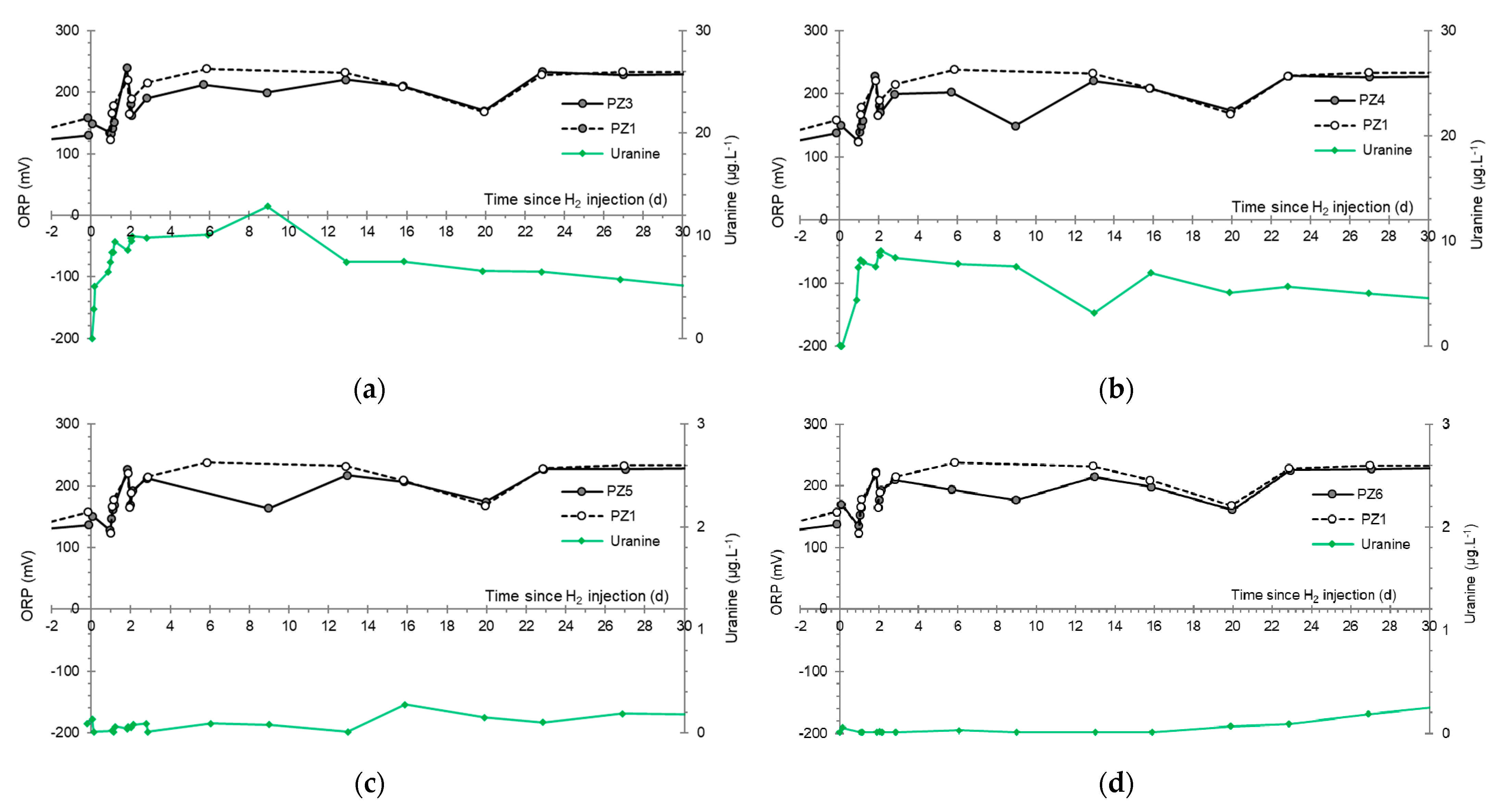
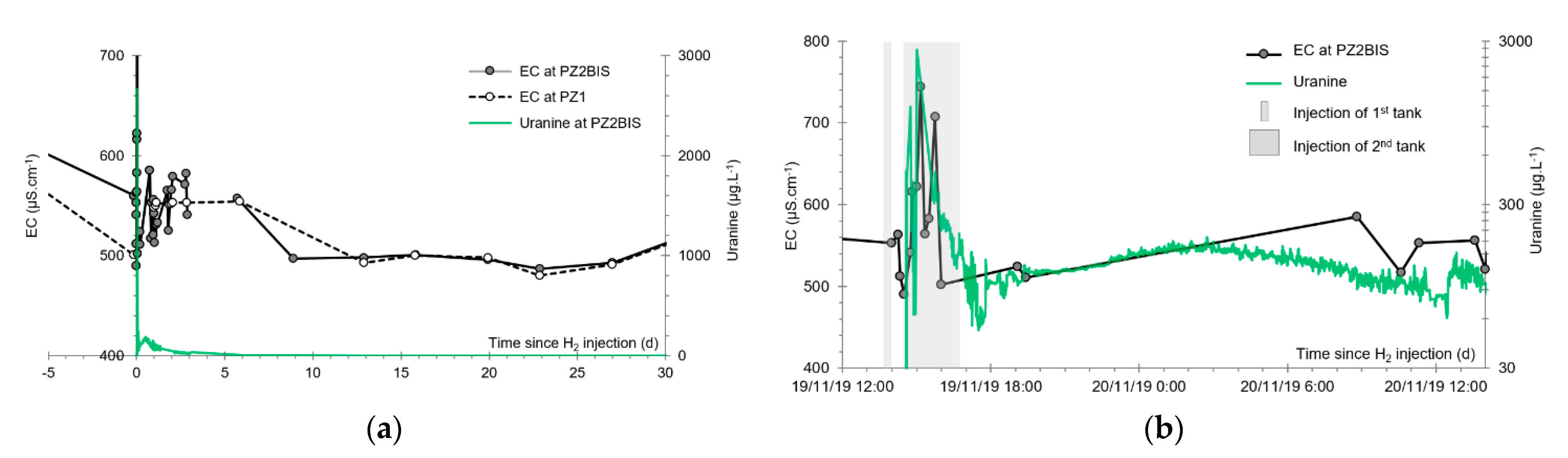
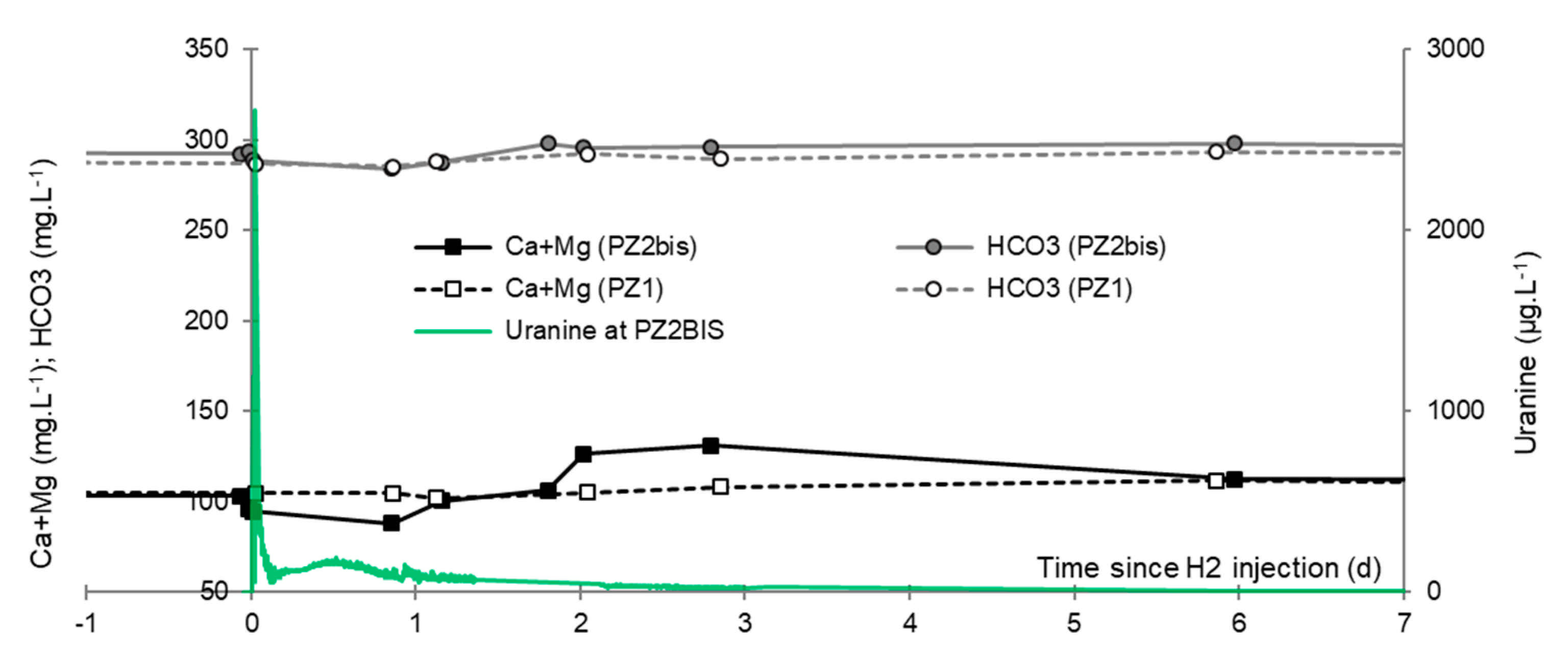
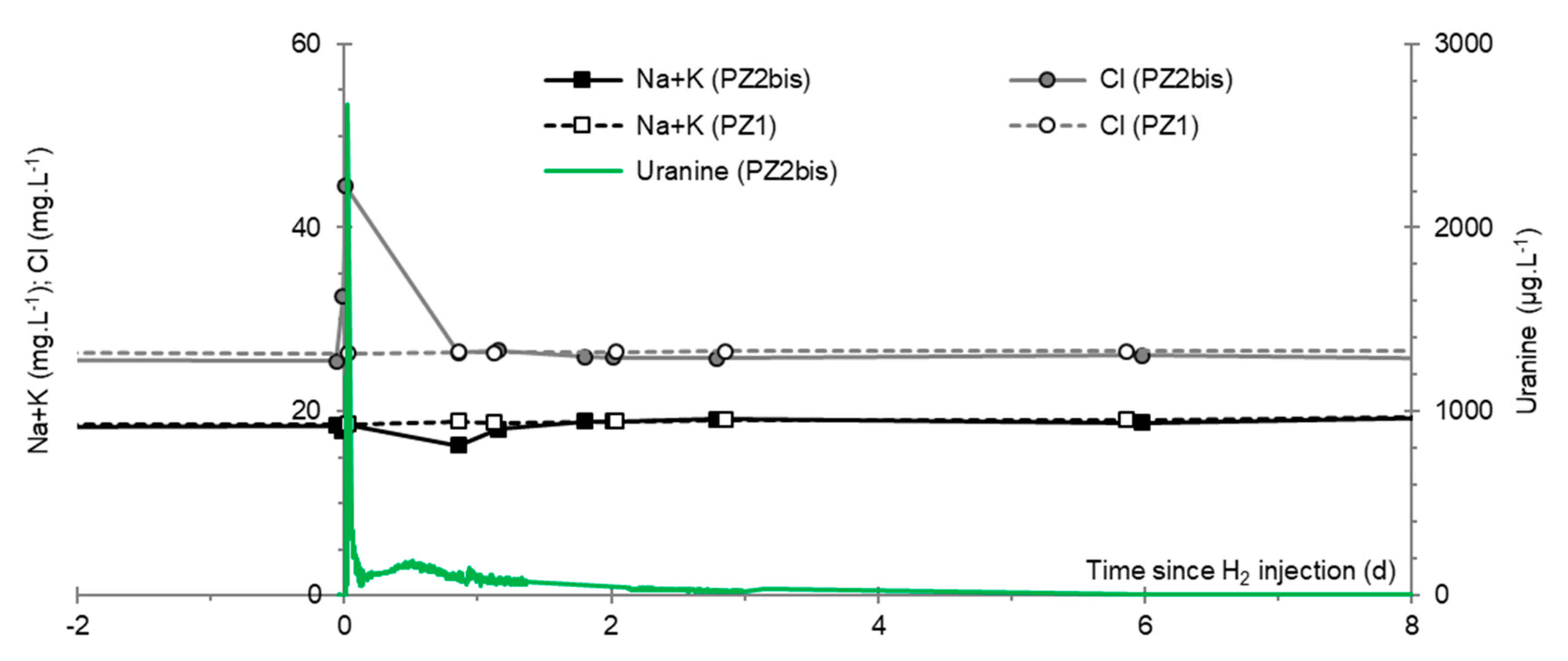
3.2. Tracing of the Injected Water
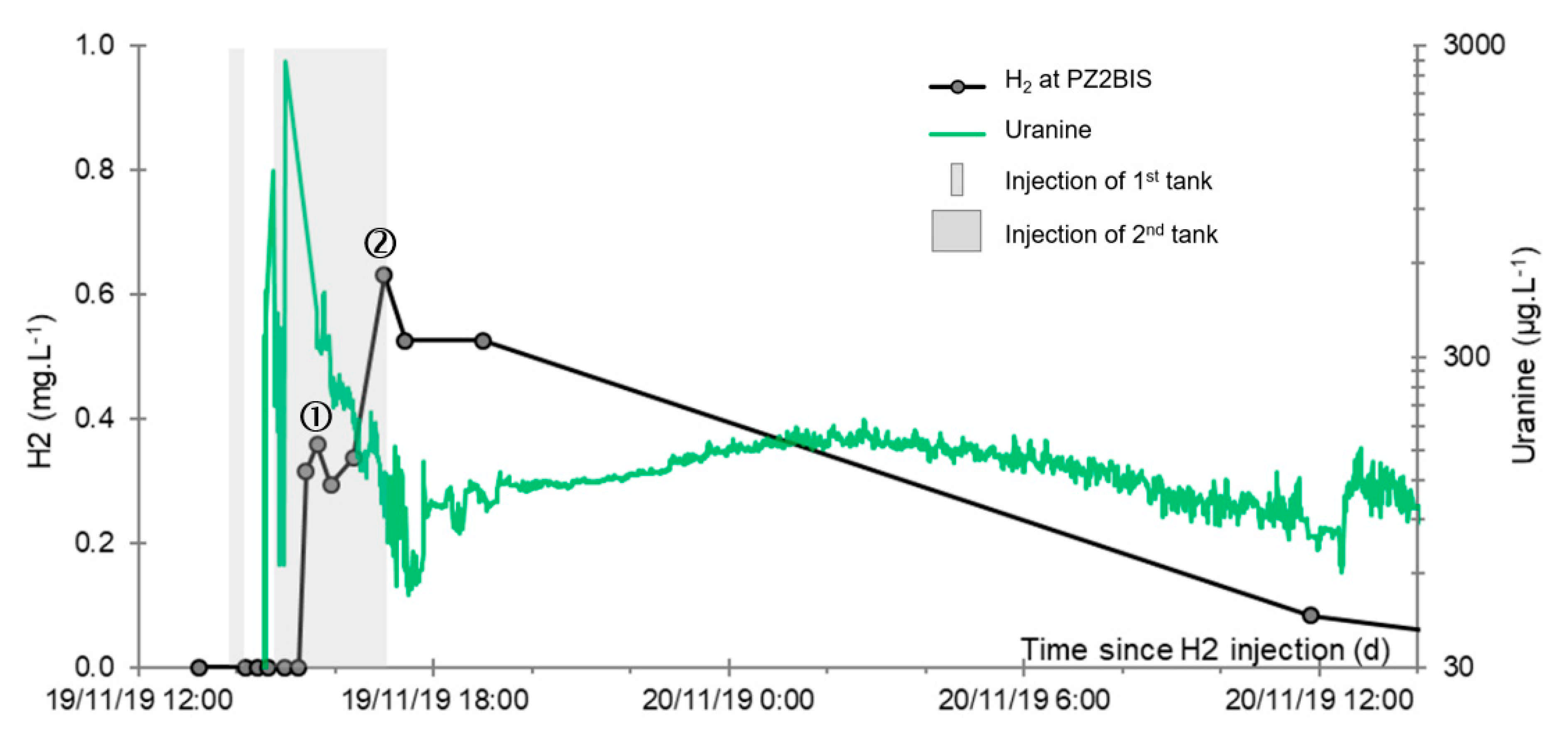
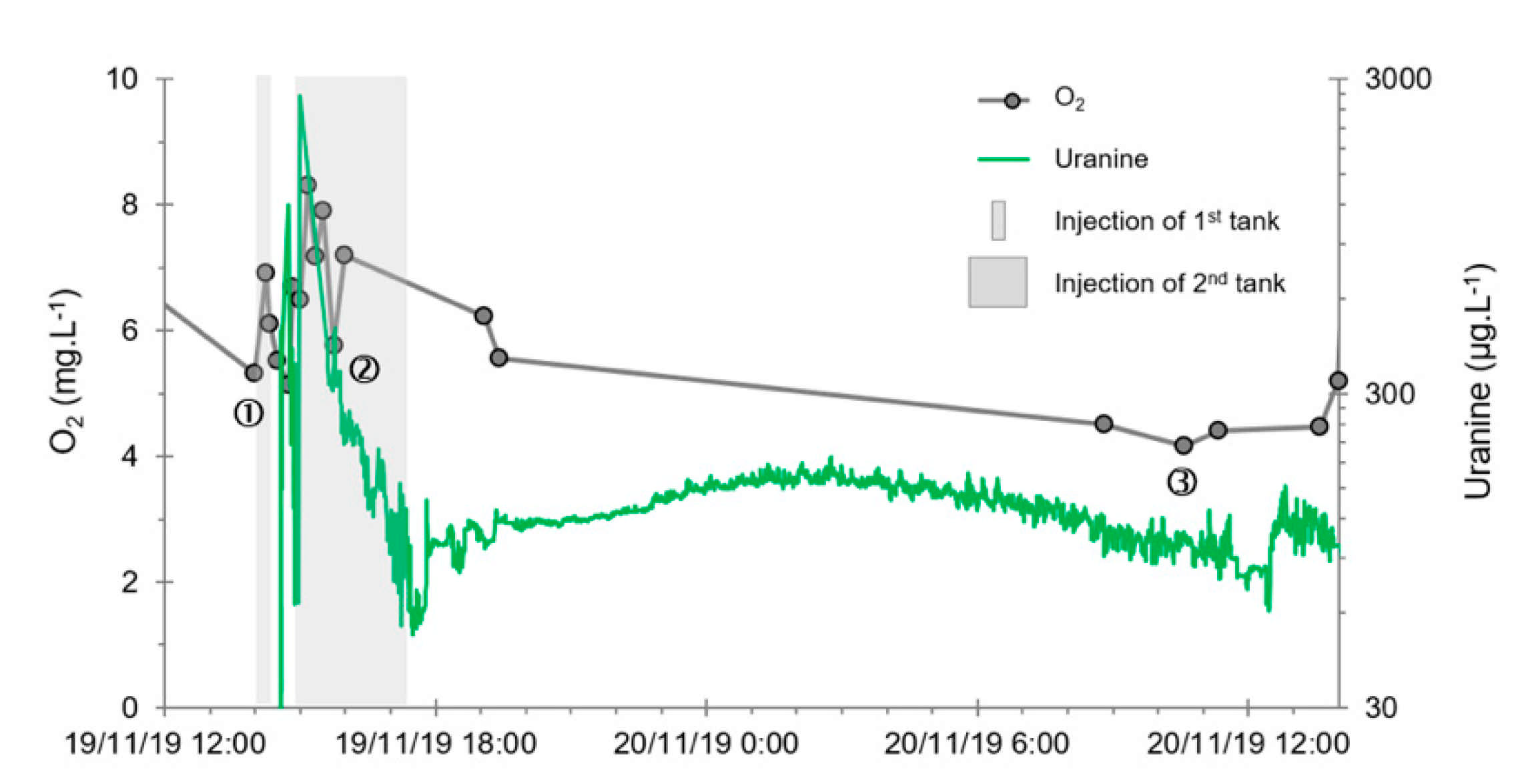
- A first plume caused a significant peak (see circled 1 in Figure 10b). It started at 14:30, i.e., 0.5 h after injection of the tracers, and peaked at 14:45 at a concentration of 1678 µg·L−1 for lithium and 1188 µg·L−1 for uranine (a signal which saturated the fluorimeters). Due to an initially inadequate measurement interval scheme, no helium was detected in this first plume that corresponds to the passage of water from the tracer tank. During this peak, which lasted approximately 1 h, the tracer concentration reached 17% of the lithium injection concentration and 12% of the uranine injection concentration, i.e., a dilution factor of 5.96 and 8.42, respectively.
- A second more intense and longer plume lasted about 3.5 h, synchronous with the injection of the hydrogenated water (see circled 2 in Figure 10b). It peaked at 15:00, i.e., approximately 10 min after the start of injection, at a concentration of 3913 µg·L−1 for lithium, 2667 µg·L−1 for uranine (again inducing saturation of the fluorimeters), and 160 µg·L−1 for helium. The tracer dilution factors reached 2.56, 3.75, and 9.28, respectively. This plume is interpreted as the result of leaching of the tracers contained in the porous matrix of the aquifer rock induced by the arrival of water at a slight overpressure from the second tank.
- In the third, weaker and more spread-out plume (see ③ in Figure 10b; note the logarithmic axes), uranine was detected by the fluorimeter. It peaked at 189 µg·L−1 on 20 November 2019 at 02:45 (t = 0.531 days) and decreased very slowly, finally lasting about a month; 0.24 µg·L−1 of uranine remained at t = 26.8 days, lithium and helium having disappeared by t = 12.9 days. When the concentration peak passed through, the dilution factor reached 52.9 for uranine, the only tracer measured using the recording fluorimeter (because the peak occurred in the middle of the night), which must have corresponded to a dilution factor of about 36 for lithium and about 130 for helium.
3.3. Dissolved Hydrogen
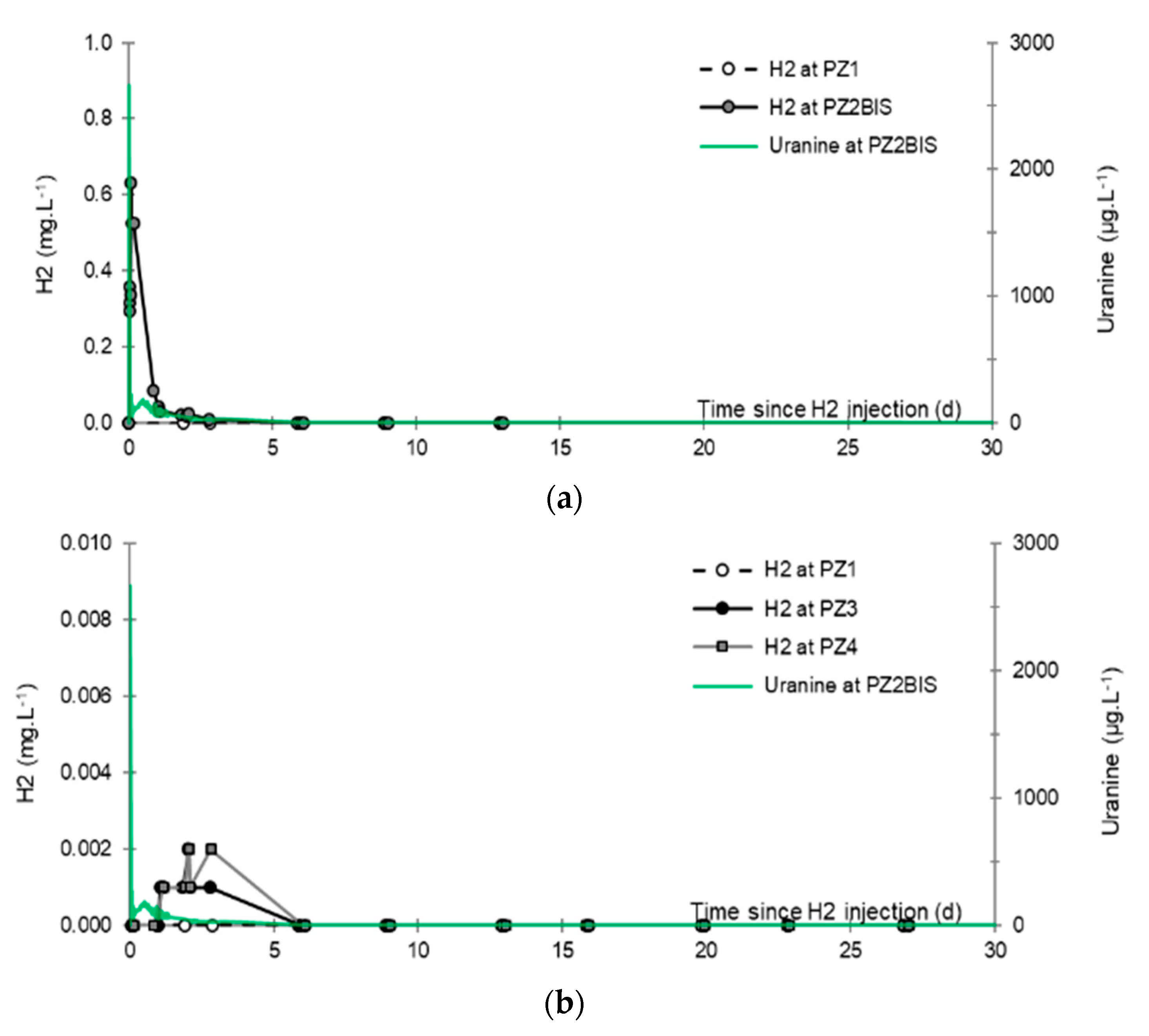
- No trace of dissolved CH4 or H2S is detected in any piezometer using the method of partial degassing by mechanical agitation.
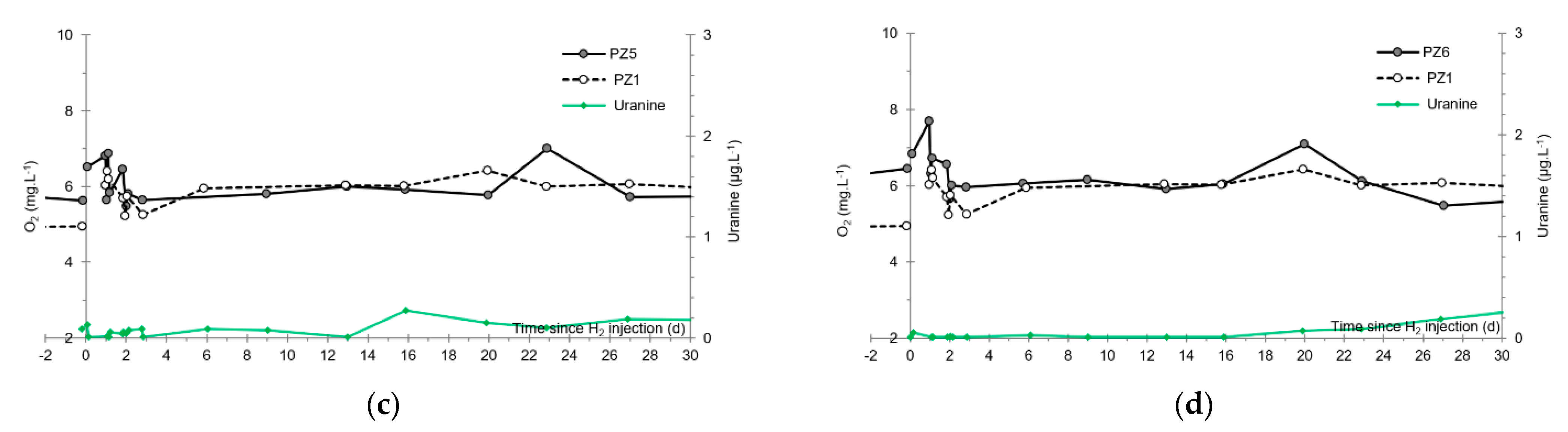
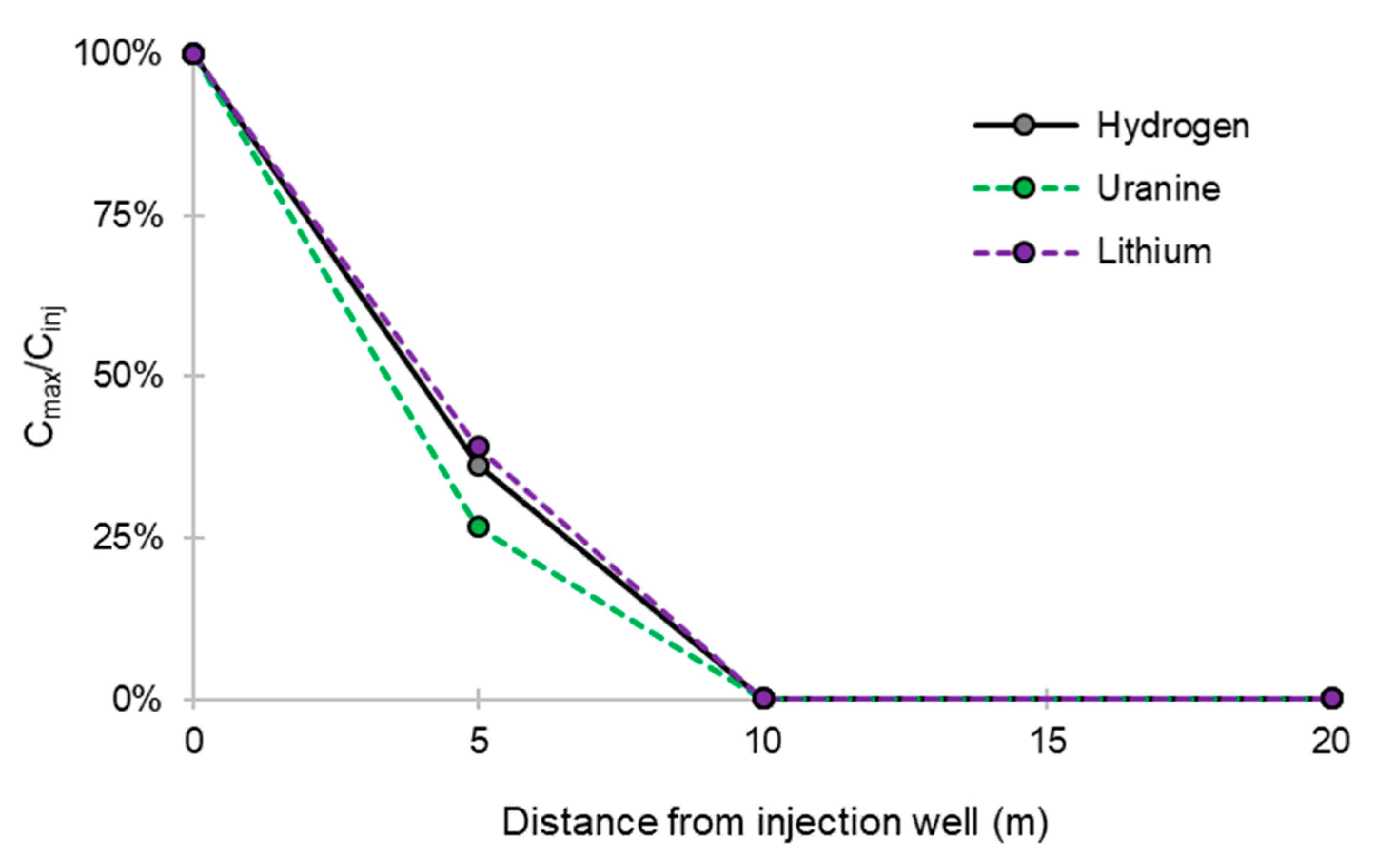
- No trace of dissolved H2 is detected at PZ1 located 20 m upstream of the injection well or at PZ5 and PZ6, respectively, located 30 and 60 m downstream of the injection well.
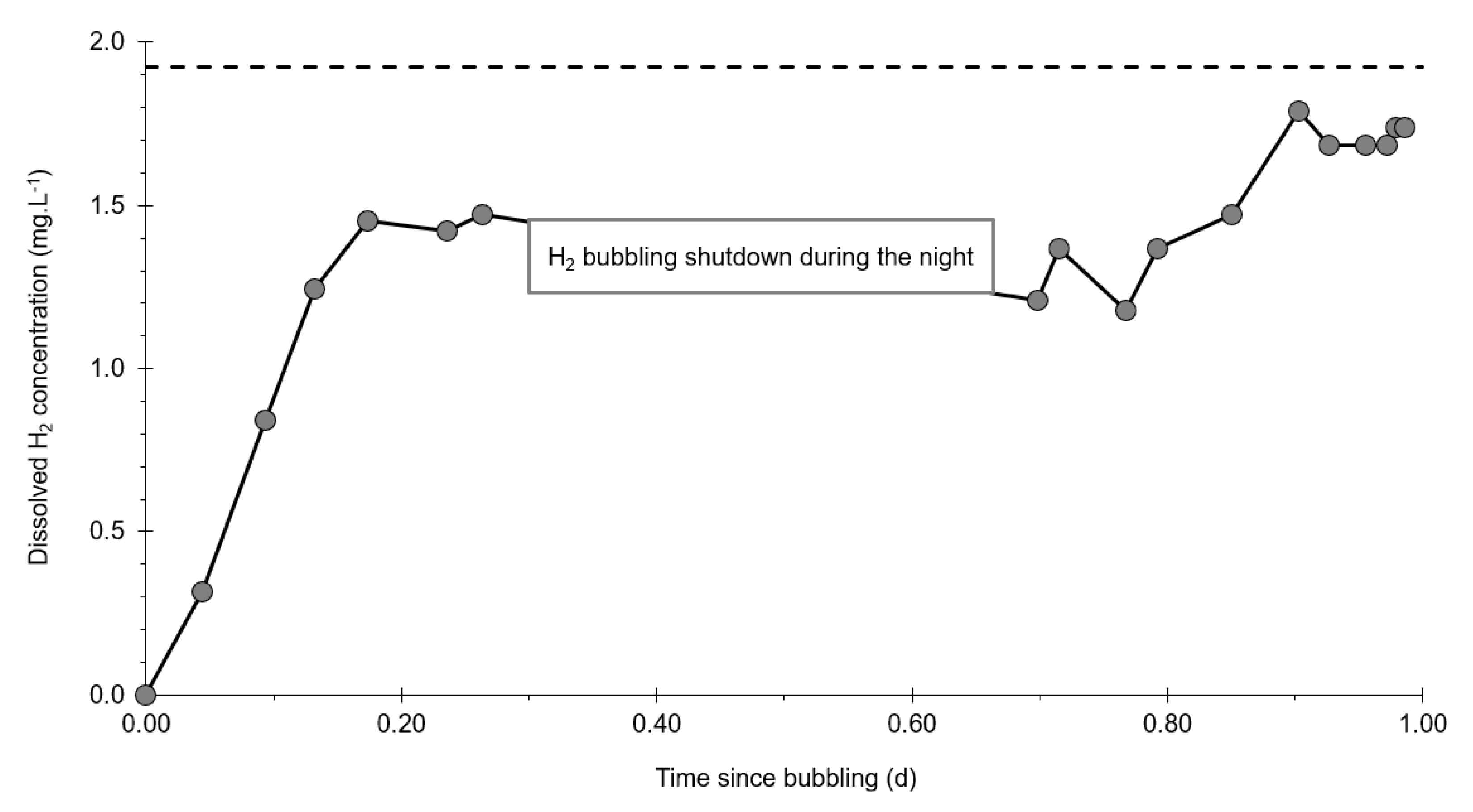
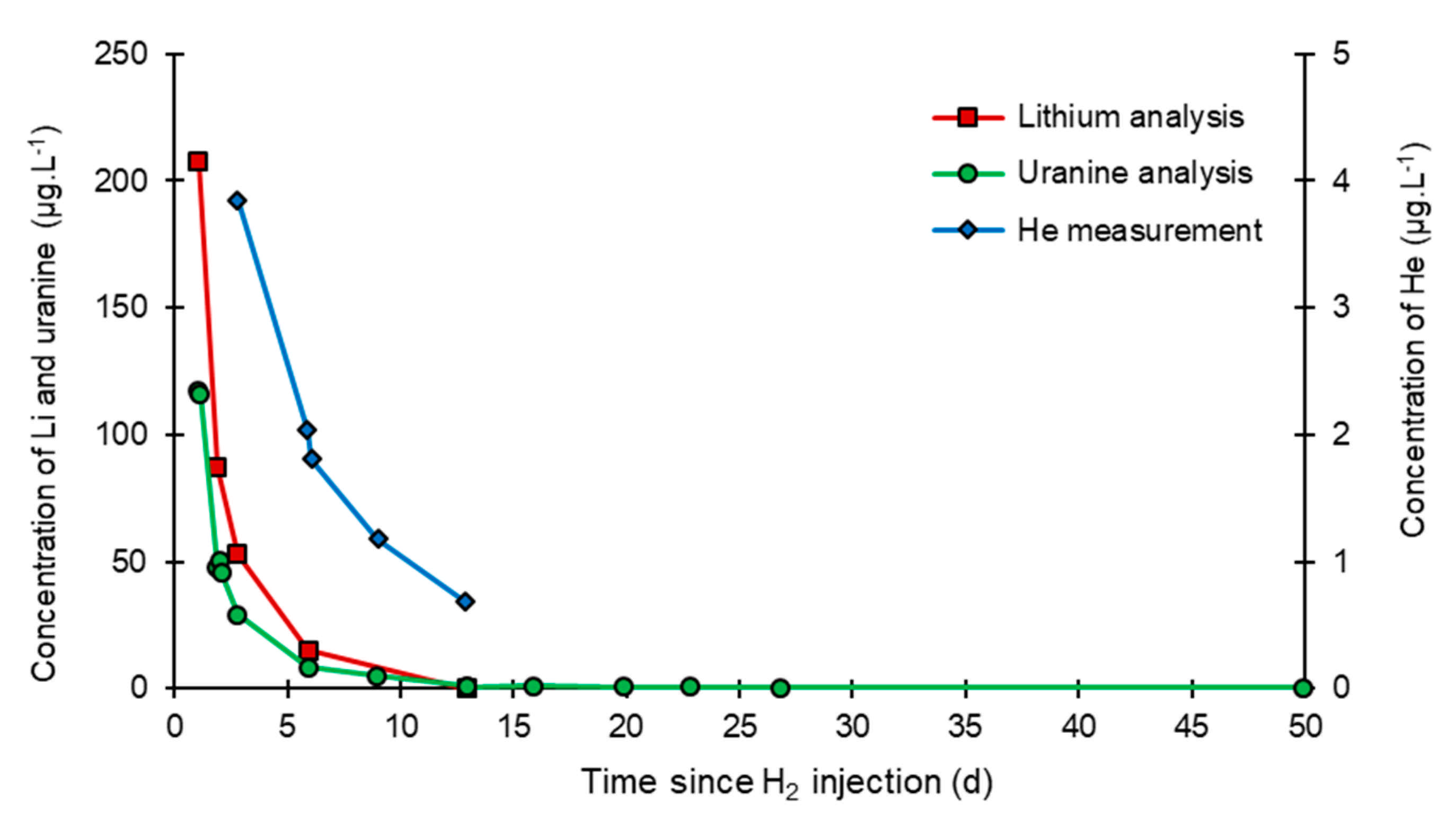
- At the injection well (PZ2), the dissolved H2 concentration, which is 1.76 mg·L−1 at the time of injection, is still 0.084 mg·L−1 when the well was reopened, i.e., 1.8 days after the start of the injection. This residual concentration represents 5% of the concentration of the injected water.
- At the main monitoring piezometer (PZ2BIS), located 5 m downstream, the concentration peak passed 2 h after the injection started (i.e., 0.08 days) with a value of 0.63 mg·L−1. This corresponds to a theoretical transfer velocity of 60 m·day−1 what is not a representative value of the mean transfer velocity of the water in the aquifer (which is approximately 3–10 m·day−1 according to Gombert et al. [7]) because it is strongly influenced by the injection.
- At the monitoring piezometer PZ2TER located 7 m downstream, the dissolved H2 concentration peak detected by Raman spectrometry passed through 9.7 h after the start of injection (i.e., 0.40 days) with a value of 0.17 mg·L−1 [25]. This corresponds to a theoretical transfer velocity of 17 m·day−1; value still strongly influenced by the injection.
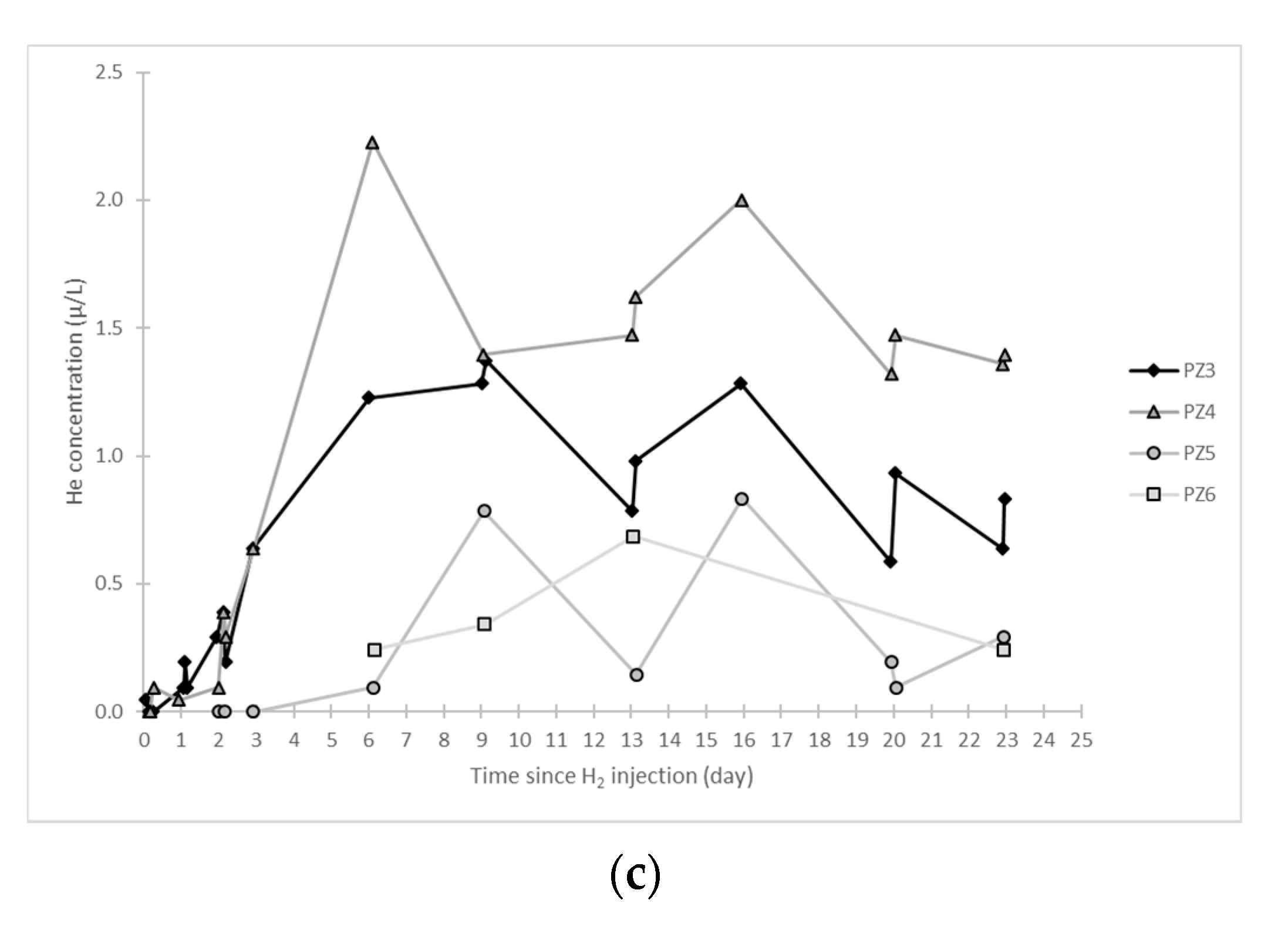
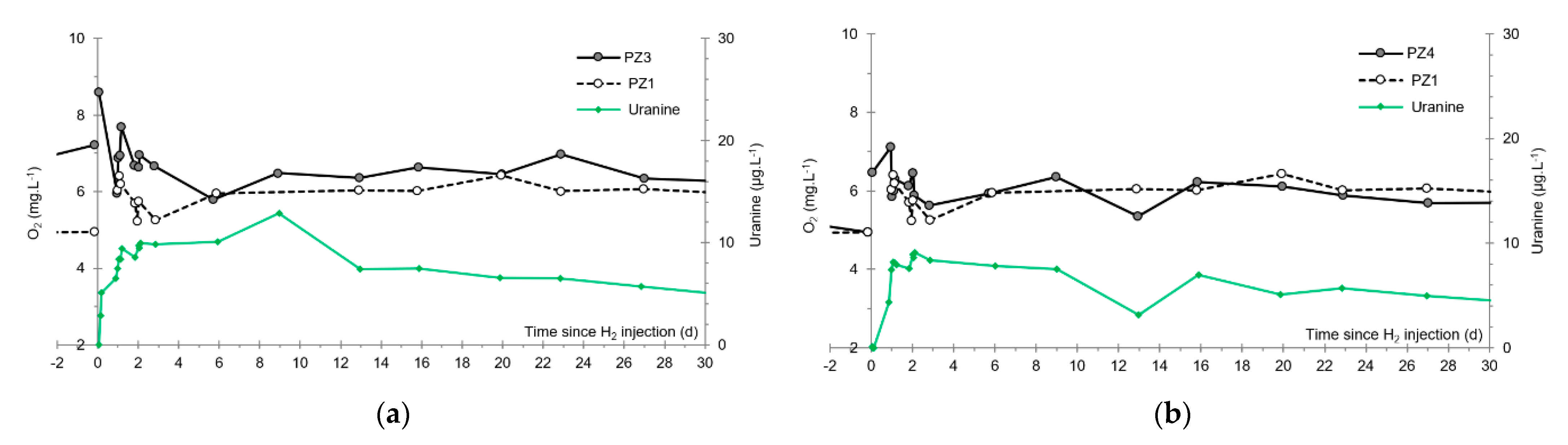
- At the PZ3 piezometer, located 10 m downstream, the first traces of dissolved H2 appeared at 1.05 days and the concentration reached its maximum of 1.7 µg·L−1 after 2.02 days, which corresponds to a transfer velocity of 5 m·day−1.
- At the PZ4 piezometer, located 20 m downstream, the first traces of dissolved H2 appeared at 1.2 days and the concentration peaked a first time at 1.5 µg·L−1 after two days and a second time at 1.74 µg·L−1 after 2.8 days. This corresponds to transfer speeds of around 10 and 7 m·day−1, respectively.
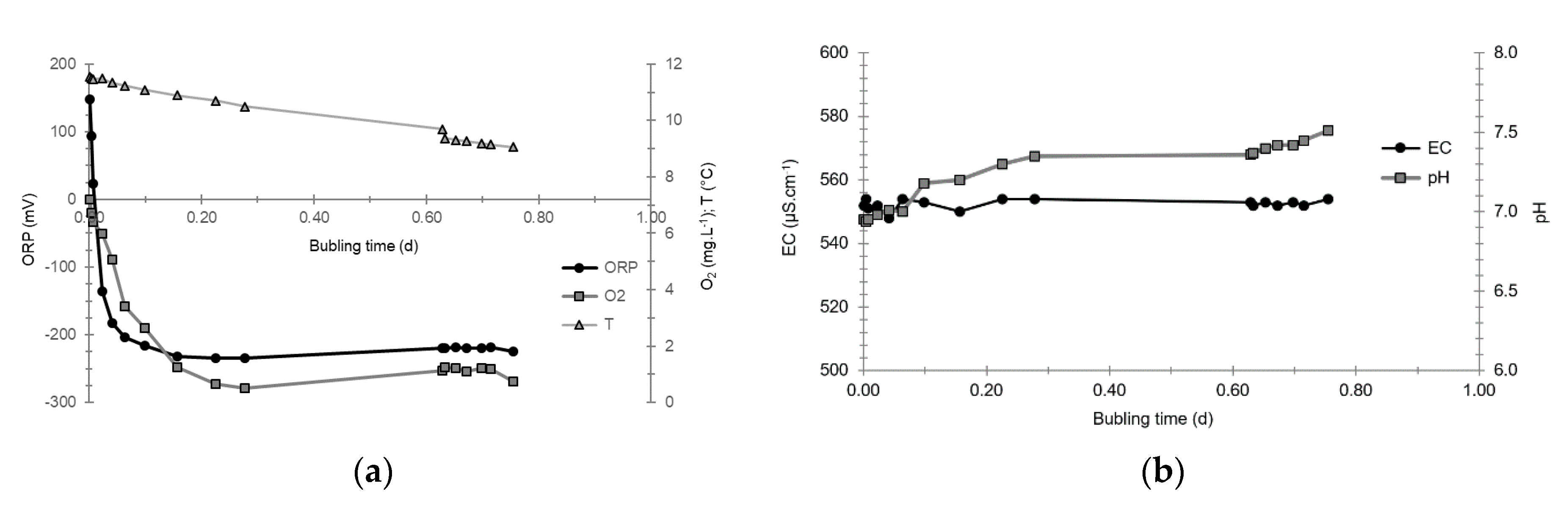
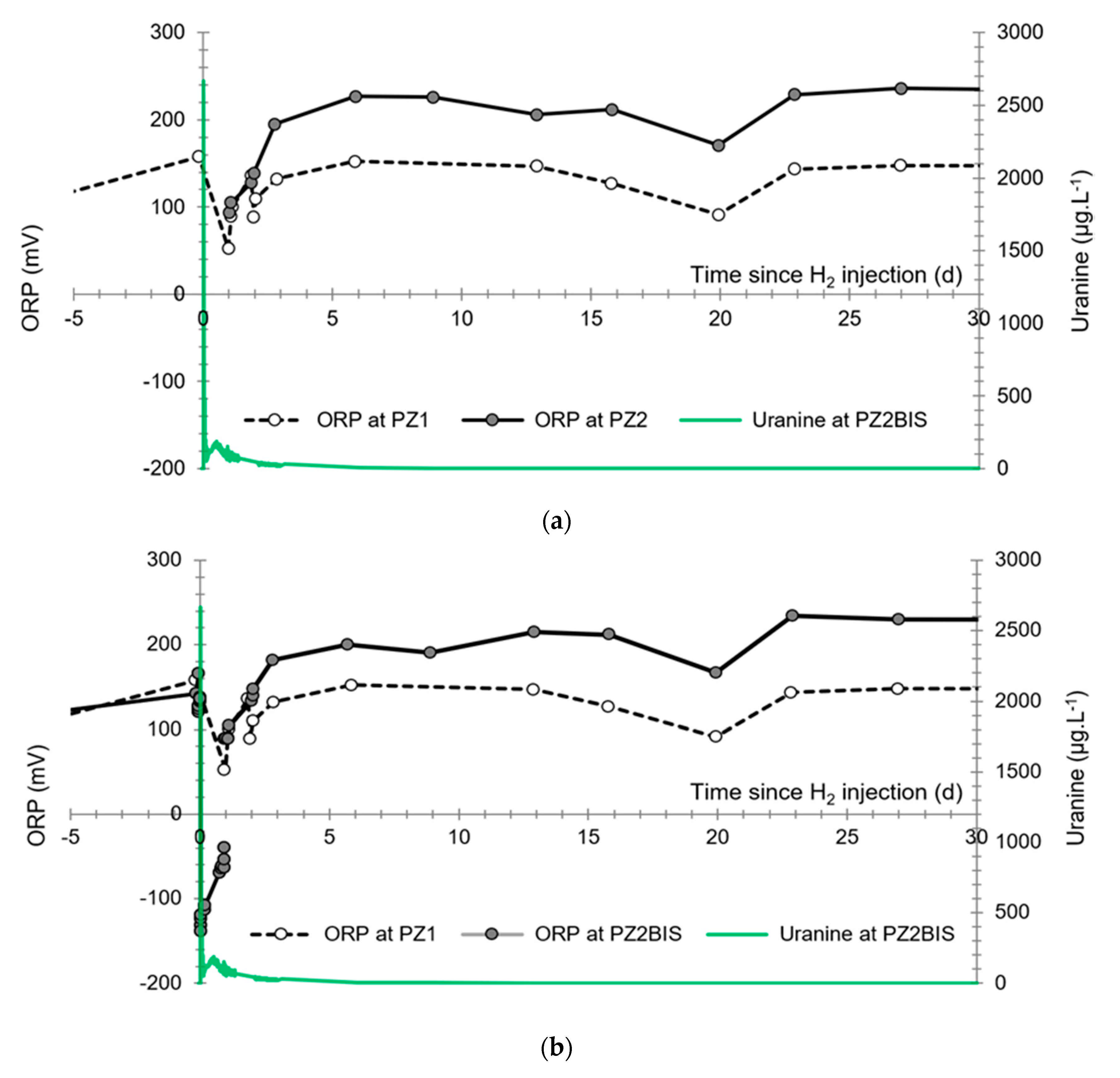
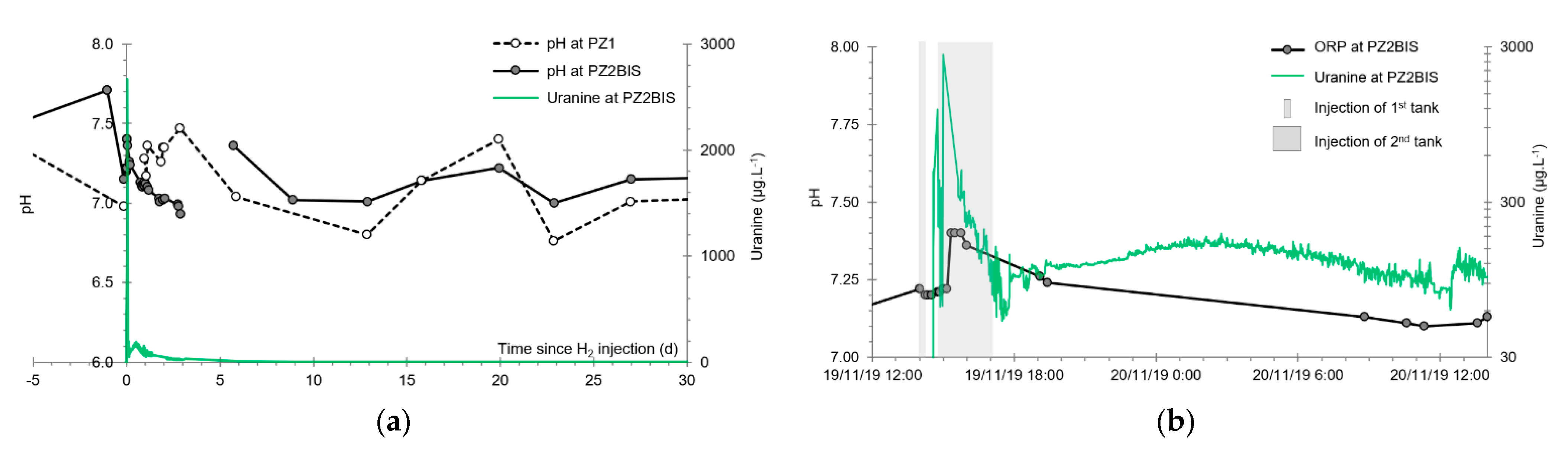
3.4. Oxidation-Reduction Potential
- The first is not very marked (−22%) and reached +120 mV at 14:15, i.e., 15 min after injection from the first tank containing helium and hydrological tracers. It corresponds to the passage of less oxidizing water, probably because of its deoxygenation induced by the introduction of dissolved helium.
- The second is very clear (−190%) and it reached −139 mV at 15:45, i.e., 55 min after injection from the second tank containing dissolved hydrogen. This drop is in fact synchronous with the second tracer peak.
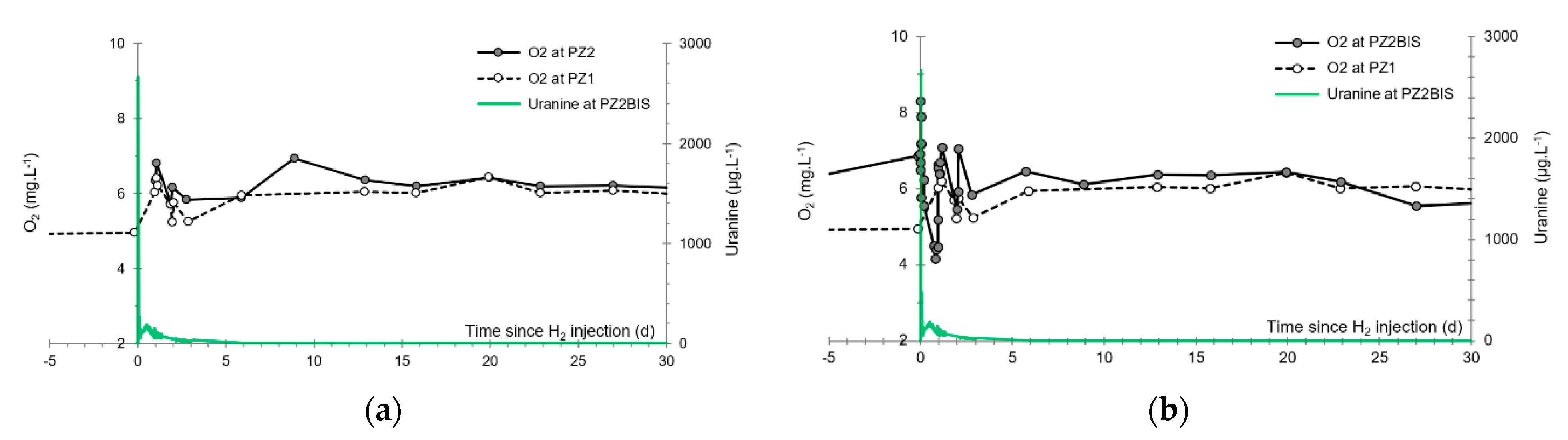
3.5. Dissolved Oxygen
- The first one, synchronous with the first tracer peak, corresponds to the passage of water from the first tracer tank (see circled 1 in Figure 18). This water is deoxygenated due to the bubbling of helium.
- The second one, synchronous with the second tracer peak, corresponds to the rapid passage of water from the second tank. This water is deoxygenated due to the bubbling of hydrogen (see circled 2 in Figure 18).
- The third one, synchronous with the third tracer peak, corresponds to the slow passage of water from the second tank (see circled 3 in Figure 18). It signals the arrival of the main plume of hydrogenated water, which circulated more slowly within the aquifer and still contained under-oxygenated water for about a day after the injections.
3.6. Other Physicochemical Parameters
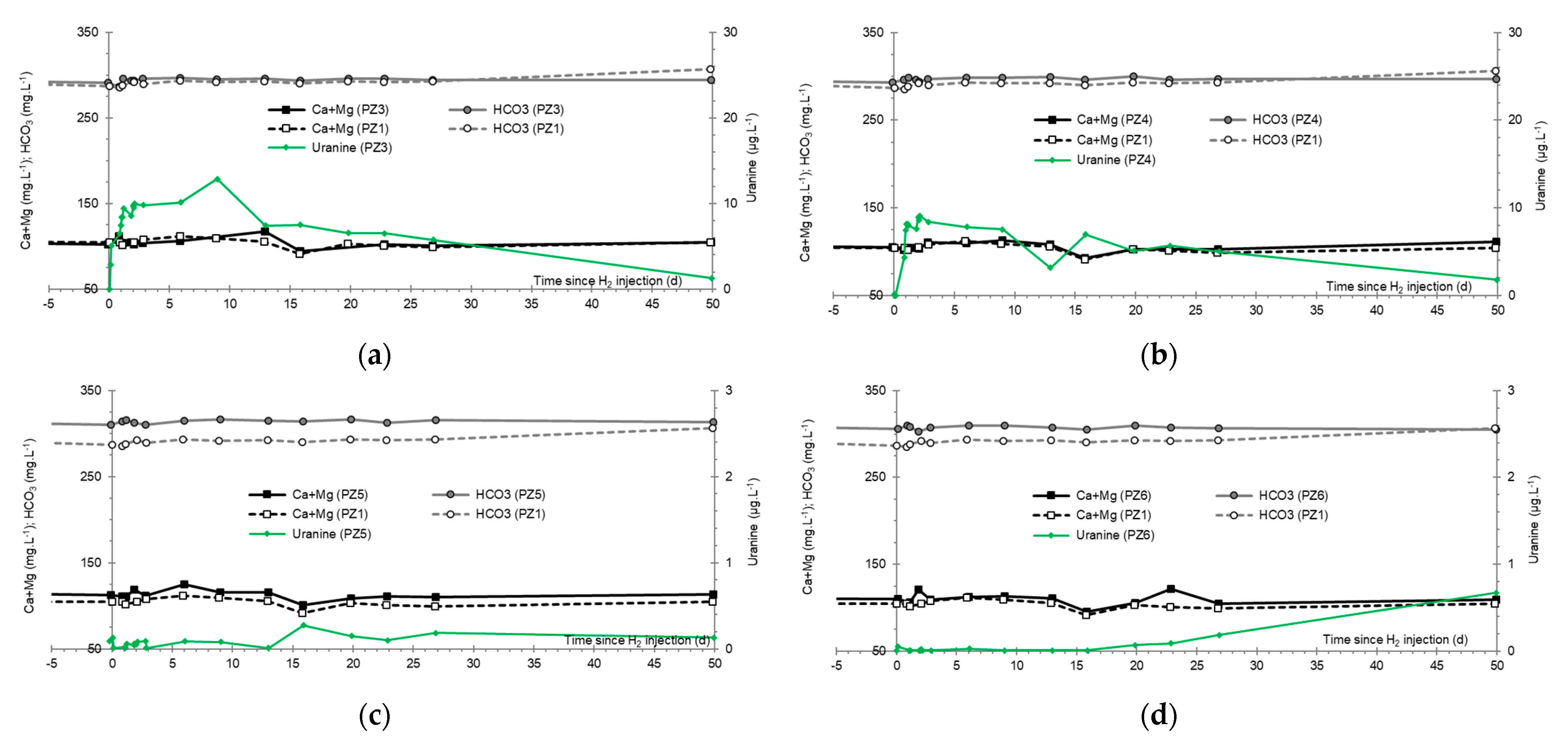
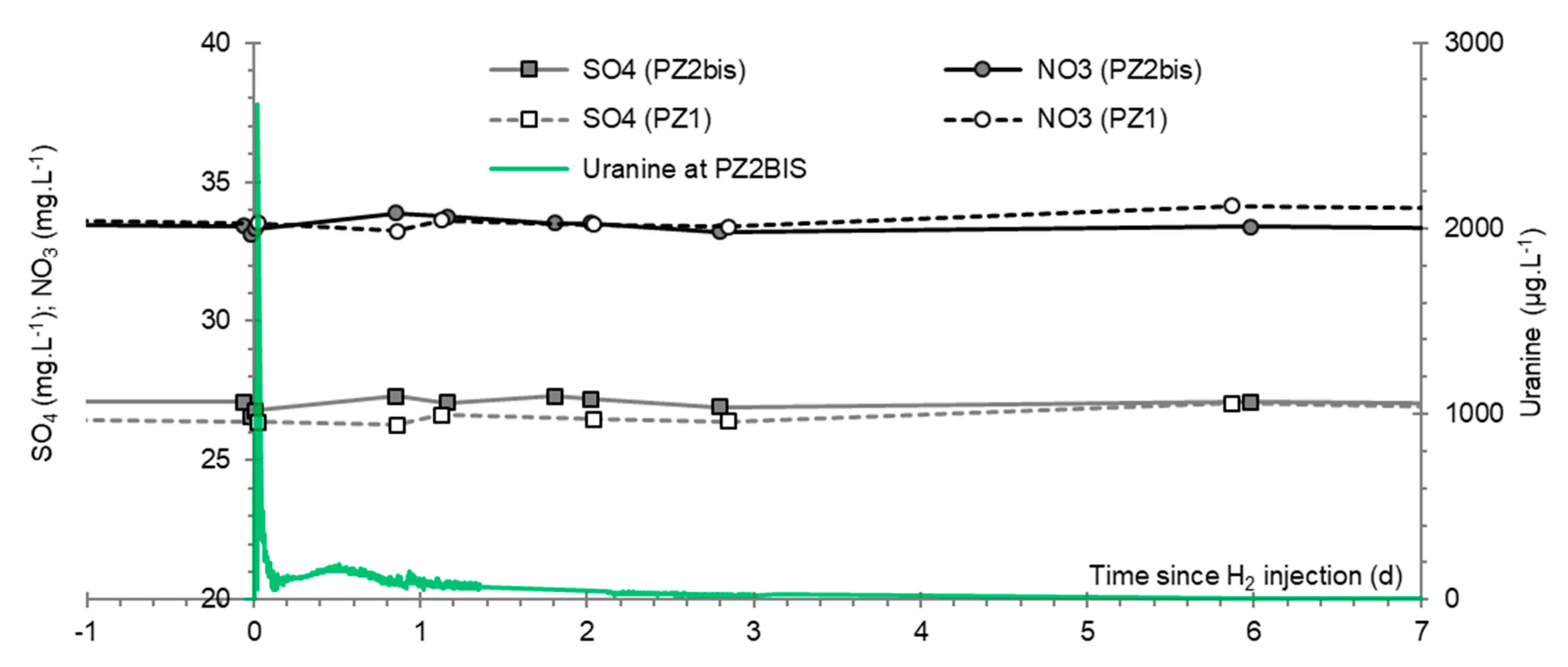
3.7. Dominant Ions (Ca2+, Mg2+ and HCO3−)
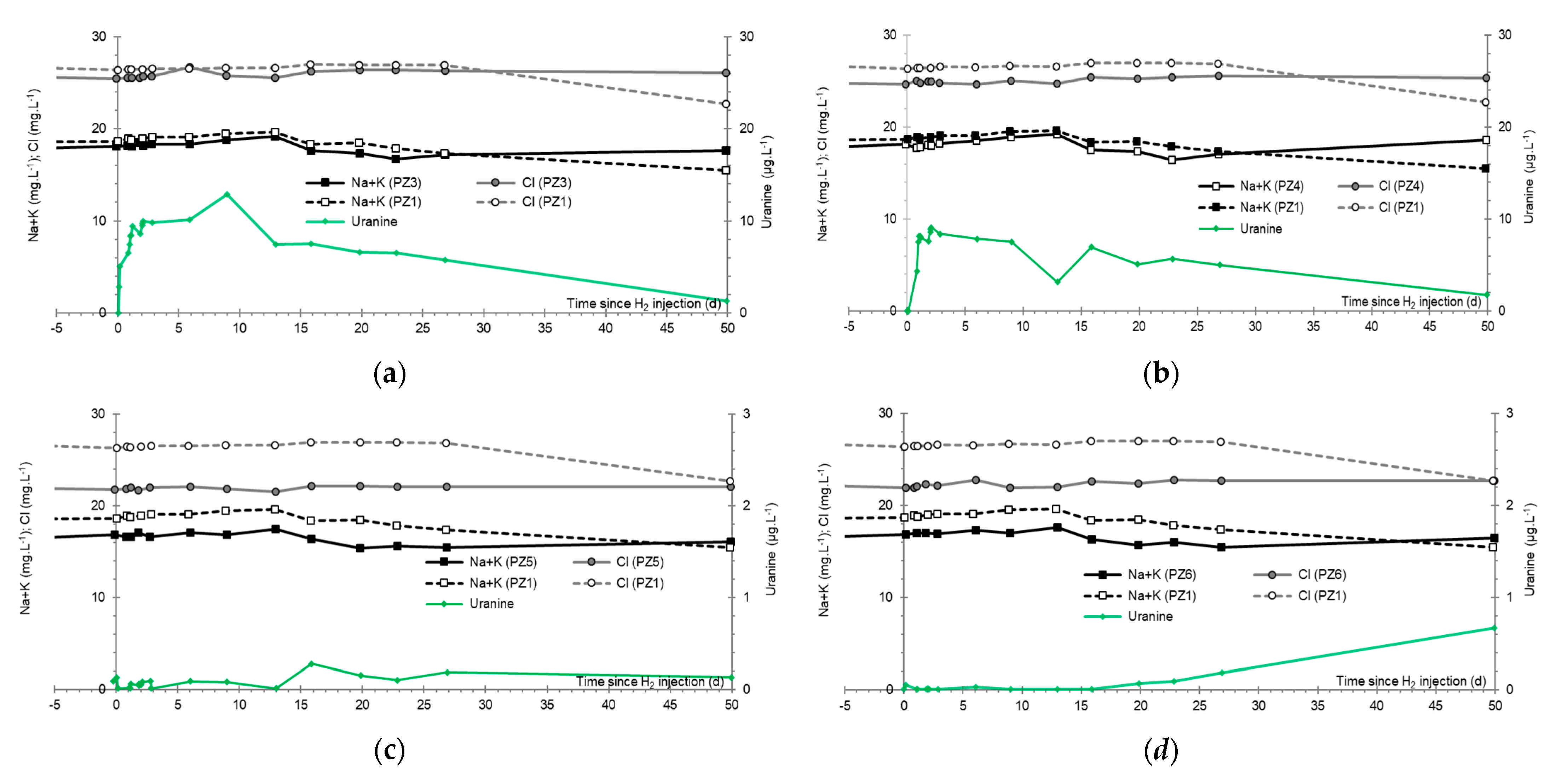
3.8. Chlor-Alkali Ions (Cl−, Na+, and K+)
3.9. Nitrates, Sulfates, and Their Derivatives
4. Discussion
5. Conclusions
- A sharp decrease of the oxidation-reduction potential
- The almost total disappearance of dissolved O2 and CO2
- A slight increase of the pH that induced the precipitation of alkaline earth bicarbonated ions and, accordingly, the decrease of electrical conductivity
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Légifrance. Loi n° 2015-992 du 17 août 2015 Relative à la Transition Energétique Pour la Croissance Verte. Available online: https://www.legifrance.gouv.fr/eli/loi/2015/8/17/DEVX1413992L/jo/texte (accessed on 25 June 2020).
- Ineris. Le Stockage Souterrain Dans le Contexte de la Transition Energétique. Maîtrise des Risques et Impacts. Ineris Références. 2016. Available online: https://www.ineris.fr/sites/ineris.fr/files/contribution/Documents/ineris-dossier-ref-stockage-souterrain.pdf (accessed on 25 June 2020).
- Panfilov, M. Underground and pipeline hydrogen storage. In Compendium of Hydrogen Energy; Gupta, R.B., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 92–116. [Google Scholar] [CrossRef]
- Caglayan, D.G.; Weber, N.; Heinrichs, H.U.; Linssen, J.; Robinius, M.; Kukla, P.A.; Stolten, D. Technical potential of salt caverns for hydrogen storage in Europe. Int. J. Hydrog. Energy 2020, 45, 6793–6805. [Google Scholar] [CrossRef]
- Lions, J.; Devau, N.; de Lary, L.; Dupraz, S.; Parmentier, M.; Gombert, P.; Dictor, M.-C. Potential impacts of leakage from CO2 geological storage on geochemical processes controlling fresh groundwater quality: A review. Int. J. Greenh. Gas Control 2014, 22, 165–175. [Google Scholar] [CrossRef]
- Lafortune, S.; Gombert, P.; Pokryszka, Z.; Lacroix, E.; de Donato, P.; Jozja, N. Monitoring scheme for the detection of hydrogen leakage from a deep underground storage. Part 1: On site validation of an experimental protocol via the combined injection of helium and tracers into an aquifer. Appl. Sci. 2020, 10, 6058. [Google Scholar] [CrossRef]
- Foh, S.; Novil, M.; Rockar, E.; Randolph, P. Underground Hydrogen Storage; Final Report, [Salt Caverns, Excavated Caverns, Aquifers and Depleted Fields]; 1979. Available online: https://www.osti.gov/servlets/purl/6536941-eQcCso/ (accessed on 22 February 2021).
- Lord, A.S. Overview of Geologic Storage of Natural Gas with an Emphasis on Assessing the Feasibility of Storing Hydrogen; Sandia National Laboratories: Albuquerque, NM, USA, 2009. [Google Scholar] [CrossRef]
- Lassin, A.; Dymitrowska, M.; Azaroual, M. Hydrogen solubility in pore water of partially saturated argillites: Application to Callovo-Oxfordian clayrock in the context of a nuclear waste geological disposal. Phys. Chem. Earth 2011. [Google Scholar] [CrossRef]
- Gombert, P.; Pokryszka, Z.; Lafortune, S.; Lions, J.; Grellier, S.; Prevot, F.; Squarcioni, P. Selection, instrumentation and characterization of a pilot site for CO2 leakage experimentation in a superficial aquifer. Energy Procedia 2014. [Google Scholar] [CrossRef]
- Gal, F.; Lions, J.; Pokryszka, Z.; Gombert, P.; Grellier, S.; Prévot, F.; Squarcioni, P. CO2 leakage in a shallow aquifer—Observed changes in case of small release. Energy Procedia 2014. [Google Scholar] [CrossRef]
- Truche, L.; Jodin-Caumon, M.C.; Lerouge, C.; Berger, G.; Mosser-Ruck, R.; Giffaut, E.; Michau, N. Sulphide mineral reactions in clay-rich rock induced by high hydrogen pressure. Application to disturbed or natural settings up to 250 °C and 30 bar. Chem. Geol. 2013, 351, 217–228. [Google Scholar] [CrossRef]
- Truche, L.; Berger, G.; Destrigneville, C.; Pages, A.; Guillaume, D.; Giffaut, E.; Jacquot, E. Experimental reduction of aqueous sulphate by hydrogen under hydrothermal conditions: Implication for the nuclear waste storage. Geochim. Cosmochim. Acta 2009, 73, 4824–4835. [Google Scholar] [CrossRef]
- Truche, L.; Berger, G.; Destrigneville, C.; Guillaume, D.; Giffaut, E. Kinetics of pyrite to pyrrhotite reduction by hydrogen in calcite buffered solutions between 90 and 180 °C: Implications for nuclear waste disposal. Geochim. Cosmochim. Acta 2010, 74, 2894–2914. [Google Scholar] [CrossRef]
- Siantar, D.P.; Schreier, C.G.; Chou, C.-S.; Reinhard, M. Treatment of 1,2-dibromo-3-chloropropane and nitrate-contaminated water with zero-valent iron or hydrogen/palladium catalysts. Water Res. 1996, 30, 2315–2322. [Google Scholar] [CrossRef]
- Smirnov, A.; Hausner, D.; Laffers, R.; Strongin, D.R.; Schoonen, M.A.A. Abiotic ammonium formation in the presence of Ni-Fe metals and alloys and its implication for the Hadean nitrogen cycle. Geochem. Trans. 2008. [Google Scholar] [CrossRef] [PubMed]
- Truche, L.; Berger, G.; Albrecht, A.; Domergue, L. Abiotic nitrate reduction induced by carbon steel and hydrogen: Implications for environmental processes in waste repositories. Appl. Geochem. 2013, 28, 155–163. [Google Scholar] [CrossRef]
- Truche, L.; Berger, G.; Albrecht, A.; Domergue, L. Engineered materials as potential geocatalysts in deep geological nuclear waste repositories: A case study of the stainless steel catalytic effect on nitrate reduction by hydrogen. Appl. Geochem. 2013, 35, 279–288. [Google Scholar] [CrossRef]
- Pintar, A.; Batista, J.; Levec, J.; Kajiuchi, T. Kinetics of the catalytic liquid-phase hydrogenation of aqueous nitrate solutions. Appl. Catal. B Environ. 1996, 11, 81–98. [Google Scholar] [CrossRef]
- Pintar, A.; Setinc, M.; Levec, J. Hardness and Salt Effects on Catalytic Hydrogenation of Aqueous Nitrate Solutions. J. Catal. 1998, 174, 72–87. [Google Scholar] [CrossRef]
- Bullister, J.L.; Guinasso, N.L., Jr.; Schink, D.R. Dissolved Hydrogen, Carbon Monoxide, and Methane at the CEPEX Site. J. Geophys. Res. 1982, 87, 2022–2034. [Google Scholar] [CrossRef]
- Berta, M.; Dethlefsen, F.; Ebert, M.; Schäfer, D.; Dahmke, A. Geochemical Effects of Millimolar Hydrogen Concentrations in Groundwater: An Experimental Study in the Context of Subsurface Hydrogen Storage. Environ. Sci. Technol. 2018, 52, 4937–4949. [Google Scholar] [CrossRef] [PubMed]
- Lagmöller, L.; Dahmke, A.; Ebert, M.; Metzgen, A.; Schäfer, D.; Dethlefsen, F. Geochemical Effects of Hydrogen Intrusions into Shallow Groundwater—An Incidence Scenario from Underground Gas Storage. Groundwater Quality 2019, Liège (B). 2019. Available online: https://www.uee.uliege.be/cms/c_4476800/fr/presentations-et-posters-gq2019 (accessed on 9 December 2019).
- Légifrance. Arrêté du 11 Janvier 2007 Relatif aux Limites et Références de Qualité Des Eaux Brutes et Des Eaux Destinées à la Consommation Humaine Mentionnées aux Articles R. 1321-2, R. 1321-3, R. 1321-7 et R. 1321-38 du Code de la Santé Publique. Available online: https://www.legifrance.gouv.fr/affichTexte.do?cidTexte=JORFTEXT000000465574 (accessed on 9 December 2019).
- Lacroix, E.; Lafortune, S.; de Donato, P.; Gombert, P.; Pokryszka, Z.; Liu, X.; Barres, O. Metrological assessment of on-site geochemical monitoring methods within an aquifer applied to the detection of H2 leakages from deep underground storages. AGU American Geophysical Union-Fall Meeting 2020, AGU, Virtual Event. 2020. Available online: https://search.proquest.com/openview/509d896f368e2b1f1c649ae4fc9dcb22/1?pq-origsite=gscholar&cbl=4882998 (accessed on 22 February 2021).
- Ineris. E.Cenaris. Cloud Monitoring Solution for Observational Research and Monitoring Services Related to Underground Operations and Geostructures. Technical Sheet. 2018. Available online: https://cenaris.ineris.fr/SYTGEMweb/public/FichesProduit/FP-ficheA3_e-cenaris-def-an/FP-ficheA3_e-cenaris-def-an.html (accessed on 28 August 2020).
- Labat, N.; Lescanne, M.; Hy-Billiot, J.; de Donato, P.; Cosson, M.; Luzzato, T.; Legay, P.; Mora, F.; Pichon, C.; Cordier, J.; et al. Carbon Capture and Storage: The Lacq Pilot (Project and Injection Period 2006–2013). Chap. 7: Environmental Monitoring and Modelling. 2015. Available online: https://www.globalccsinstitute.com/archive/hub/publications/194253/carbon-capture-storage-lacq-pilot.pdf (accessed on 13 December 2019).
- Cai, S.; González-Vila, Á.; Zhang, X.; Guo, T.; Caucheteur, C. Palladium-coated plasmonic optical fiber gratings for hydrogen detection. Opt. Lett. 2019, 44, 4483–4486. [Google Scholar] [CrossRef] [PubMed]
- Brouyère, S. Modelling the migration of contaminants through variably saturated dual-porosity, dual-permeability chalk. J. Contam. Hydrol. 2006, 2, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Gombert, P. Proposition de protocole de traçage appliqué au karst de la craie. Eur. J. Water Qual. 2007, 38, 61–78. [Google Scholar] [CrossRef]































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Impacted Species | H2 Partial Pressure (bar) | Temperature (°C) | pH | Catalyst | Resulting Species | Reference |
|---|---|---|---|---|---|---|
| SO42− | 4–16 | 250–300 | ≤5.6 | None | H2S | [13] |
| FeS2 * | 8–18 | 90–180 | 6.9–8.7 | None | FeS1+x, H2S | [14] |
| FeS2 * | 3–30 | 90–250 | 7.8–9.8 | None | FeS1+x, H2S | [12] |
| NO3− | 0.1 | 22 | 7.0–8.7 | Fe | NO2− | [15] |
| NO3− | 0.05 | 200 | ~6 | Fe | NH4+ | [16] |
| NO3− | 7.5 | 90–180 | 4–9 | Carbon steel | NH4+ | [17] |
| NO3− | 0.2–7.5 | 90–150 | 4–9 | Stainless steel | NH4+ | [18] |
| NO3− | 0.1–0.5 | 7–25 | 5–11 | Pd, Cu | N2 | [19,20] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gombert, P.; Lafortune, S.; Pokryszka, Z.; Lacroix, E.; de Donato, P.; Jozja, N. Monitoring Scheme for the Detection of Hydrogen Leakage from a Deep Underground Storage. Part 2: Physico-Chemical Impacts of Hydrogen Injection into a Shallow Chalky Aquifer. Appl. Sci. 2021, 11, 2686. https://doi.org/10.3390/app11062686
Gombert P, Lafortune S, Pokryszka Z, Lacroix E, de Donato P, Jozja N. Monitoring Scheme for the Detection of Hydrogen Leakage from a Deep Underground Storage. Part 2: Physico-Chemical Impacts of Hydrogen Injection into a Shallow Chalky Aquifer. Applied Sciences. 2021; 11(6):2686. https://doi.org/10.3390/app11062686
Chicago/Turabian StyleGombert, Philippe, Stéphane Lafortune, Zbigniew Pokryszka, Elodie Lacroix, Philippe de Donato, and Nevila Jozja. 2021. "Monitoring Scheme for the Detection of Hydrogen Leakage from a Deep Underground Storage. Part 2: Physico-Chemical Impacts of Hydrogen Injection into a Shallow Chalky Aquifer" Applied Sciences 11, no. 6: 2686. https://doi.org/10.3390/app11062686
APA StyleGombert, P., Lafortune, S., Pokryszka, Z., Lacroix, E., de Donato, P., & Jozja, N. (2021). Monitoring Scheme for the Detection of Hydrogen Leakage from a Deep Underground Storage. Part 2: Physico-Chemical Impacts of Hydrogen Injection into a Shallow Chalky Aquifer. Applied Sciences, 11(6), 2686. https://doi.org/10.3390/app11062686