
Identification of Key Genes Related to the Prognosis of Esophageal Squamous Cell Carcinoma Based on Chip Re-Annotation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Data Set
2.2. Chip Re-Annotation
- Downloaded the matrix file of these unknown expression sequence tags and obtained the nucleic acid sequence of these probes;
- Removed probes matching lncRNA at the same time. A total of 28,885 reannotated mRNA probes were obtained, none of which duplicated each other.
2.3. Differential Expression Analysis and Functional Enrichment Analysis
2.4. Construction of Protein-Protein Interaction Network (PPI)
2.5. Identification of Prognostic Genes in Key Genes
2.6. Exploration of the Diagnostic Efficacy of Prognostic Genes
2.7. Cell Culture
2.8. Validation Using Independent External Database and Western Blot
3. Results
3.1. LncRNA Probe Reannotation
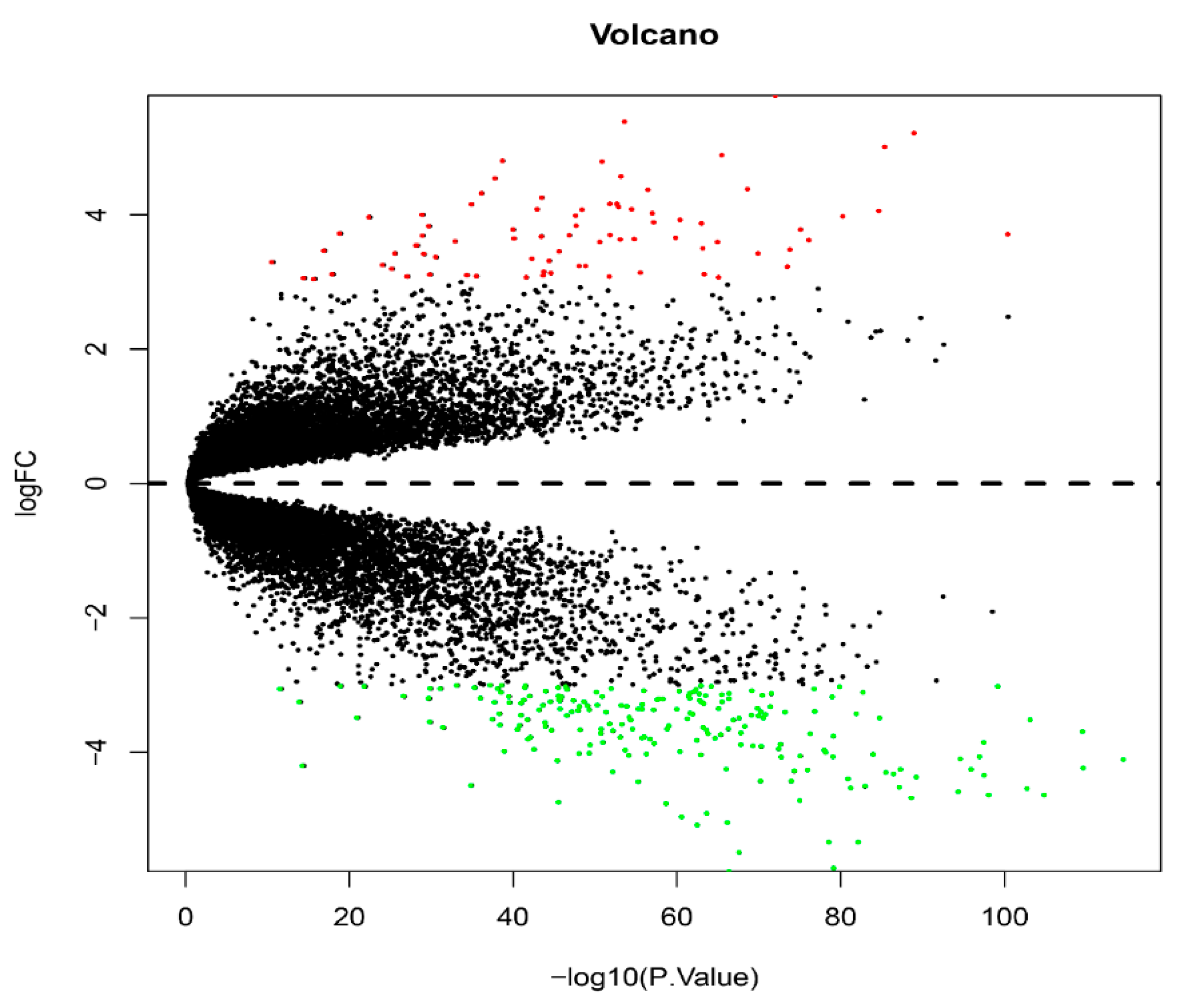
3.2. The Acquisition of Differential Genes
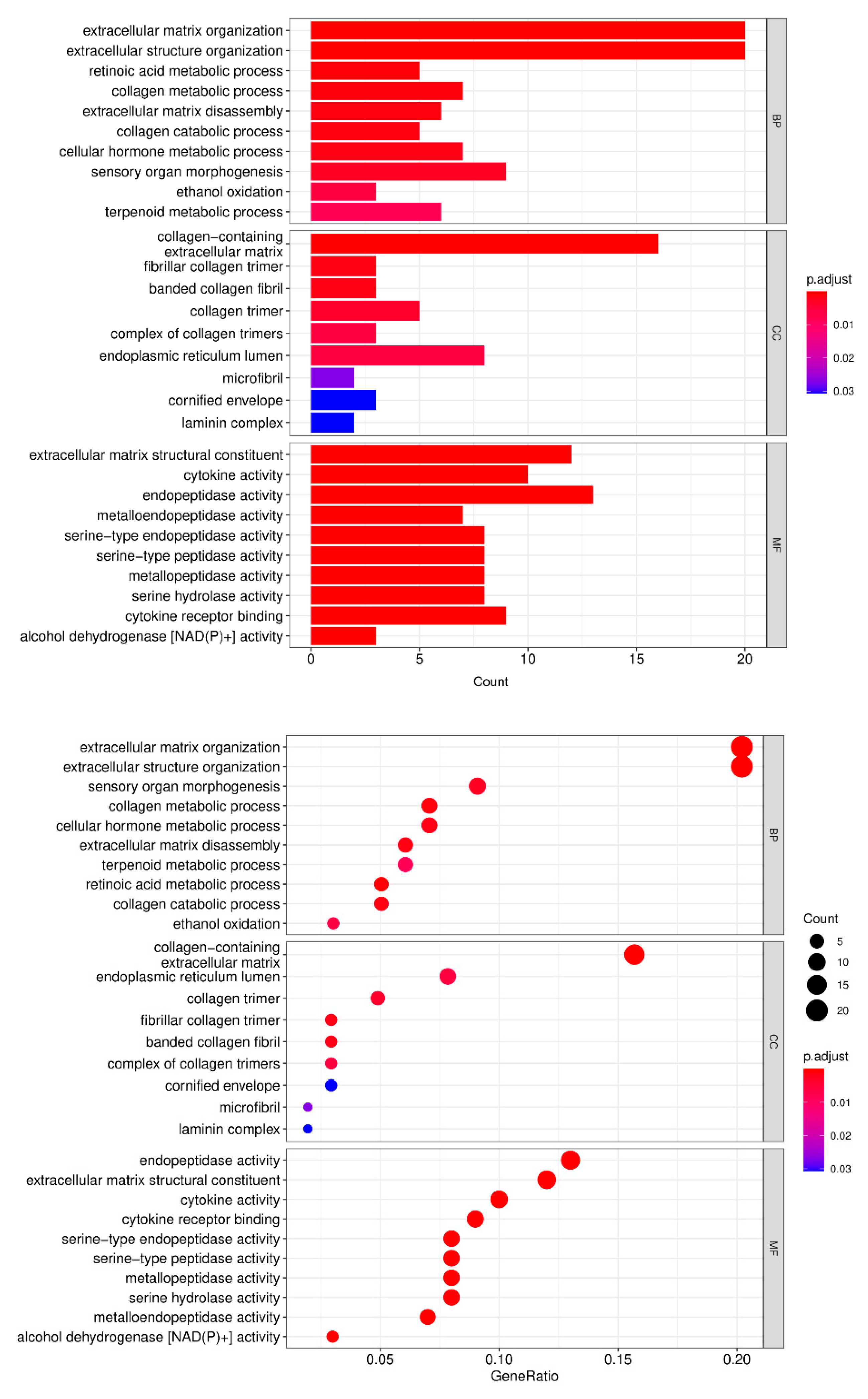
3.3. Genetic Ontology (GO) Analysis
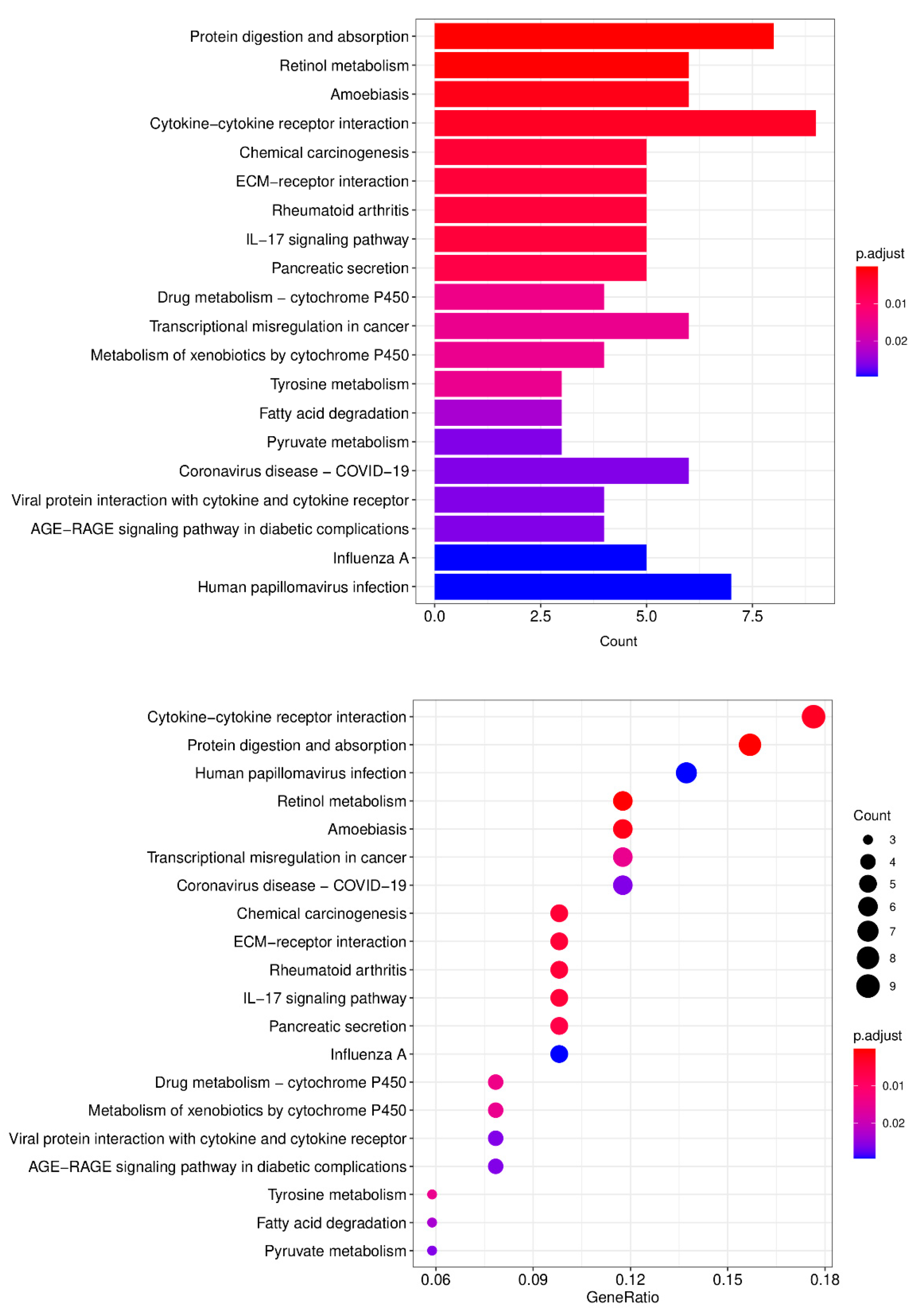
3.4. Kyoto Encyclopedia of Genes and Genome (KEGG) Pathway Enrichment Analysis
3.5. Protein–Protein Interaction Networks of DEGs
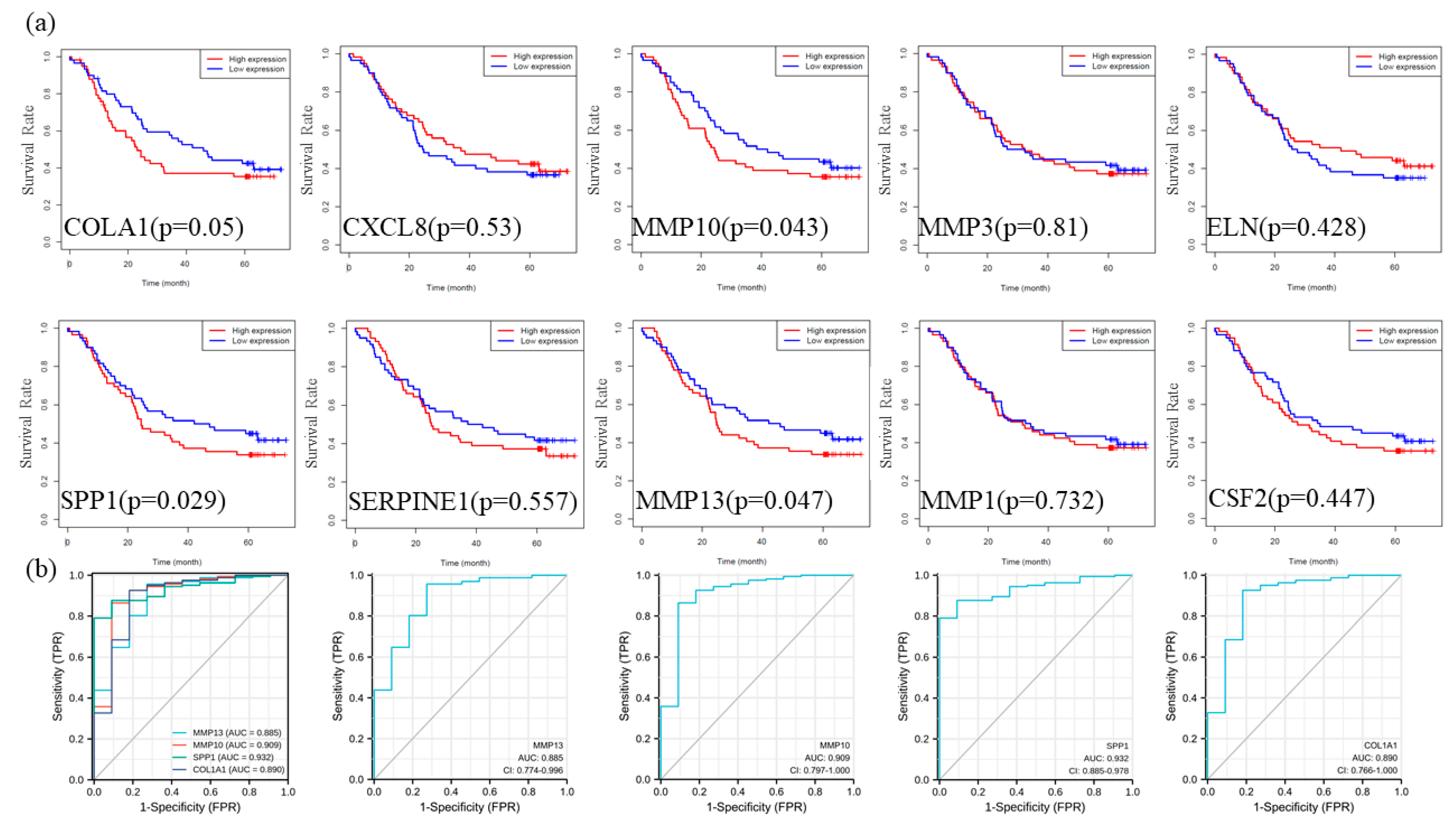
3.6. Prognostic Analysis and Diagnostic Efficacy Analysis
3.7. Validation of DEGs Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malhotra, G.K.; Yanala, U.; Ravipati, A.; Follet, M.; Vijayakumar, M.; Are, C. Global trends in esophageal cancer. J. Surg. Oncol. 2017, 115, 564–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Lu, Z.M.; Chen, K.N.; Guo, M.; Xing, H.P.; Mei, Q.; Yang, H.H.; Lechner, J.F.; Ke, Y. Human papillomavirus type 16 is an important infectious factor in the high incidence of esophageal cancer in Anyang area of China. Carcinogenesis 2001, 22, 929–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Erratum: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2020, 70, 313. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiyama, T.; Yoshihara, M.; Tanaka, S.; Chayama, K. Genetic polymorphisms and esophageal cancer risk. Int. J. Cancer 2007, 121, 1643–1658. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Rustgi, A.K.; El-Serag, H.B. Esophageal Carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef]
- Li, J.; Chen, Z.; Tian, L.; Zhou, C.; He, M.Y.; Gao, Y.; Wang, S.; Zhou, F.; Shi, S.; Feng, X.; et al. LncRNA profile study reveals a three-lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut 2014, 63, 1700–1710. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Wong, W.H. SeqMap: Mapping massive amount of oligonucleotides to the genome. Bioinformatics 2008, 24, 2395–2396. [Google Scholar] [CrossRef] [Green Version]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 4897414, 57–74. [Google Scholar] [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.F.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43. [Google Scholar] [CrossRef]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Ginestet, C. ggplot2: Elegant Graphics for Data Analysis. J. R. Stat. Soc. A Stat. 2011, 174, 245. [Google Scholar] [CrossRef]
- Yu, G.C.; Wang, L.G.; Yan, G.R.; He, Q.Y. DOSE: An R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.C.A. Modeling survival data: Extending the Cox model. Sociol. Methods Res. 2003, 32, 117–120. [Google Scholar] [CrossRef]
- De Silva, R.A.; Appulage, D.K.; Pietraszkiewicz, H.; Bobbitt, K.R.; Media, J.; Shaw, J.; Valeriote, F.A.; Andreana, P.R. The entirely carbohydrate immunogen Tn-PS A1 induces a cancer cell selective immune response and cytokine IL-17. Cancer Immunol. Immunother. 2012, 61, 581–585. [Google Scholar] [CrossRef] [Green Version]
- Kolls, J.K.; Linden, A. Interleukin-17 family members and inflammation. Immunity 2004, 21, 467–476. [Google Scholar] [CrossRef] [Green Version]
- El-Tanani, M.K. Role of osteopontin in cellular signaling and metastatic phenotype. Front. Biosci. 2008, 13, 4276–4284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudland, P.S.; Platt-Higgins, A.; El-Tanani, M.; De Silva Rudland, S.; Barraclough, R.; Winstanley, J.H.; Howitt, R.; West, C.R. Prognostic significance of the metastasis-associated protein osteopontin in human breast cancer. Cancer Res. 2002, 62, 3417–3427. [Google Scholar] [PubMed]
- Walaszek, K.; Lower, E.E.; Ziolkowski, P.; Weber, G.F. Breast cancer risk in premalignant lesions: Osteopontin splice variants indicate prognosis. Br. J. Cancer 2018, 119, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Lin, D.; Yuan, J.; Xiao, T.; Zhang, H.; Sun, W.; Han, N.; Ma, Y.; Di, X.; Gao, M.; et al. Overexpression of osteopontin is associated with more aggressive phenotypes in human non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 4646–4652. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.F.; Li, Q.H.; Zeng, L.R.; Yang, X.Y.; Peng, P.L.; He, J.H.; Fan, B. A 4-gene prognostic signature predicting survival in hepatocellular carcinoma. J. Cell. Biochem. 2019, 120, 9117–9124. [Google Scholar] [CrossRef]
- Zeng, B.; Zhou, M.; Wu, H.; Xiong, Z. SPP1 promotes ovarian cancer progression via Integrin beta1/FAK/AKT signaling pathway. OncoTargets Ther. 2018, 11, 1333–1343. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.T.; Guo, C.; Ding, X.; Fan, W.J.; Zhang, F.H.; Xu, W.L.; Ma, Y.C. Role of osteopontin in the regulation of human bladder cancer proliferation and migration in T24 cells. Mol. Med. Rep. 2015, 11, 3701–3707. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; Kundu, G.C. Osteopontin selectively regulates p70S6K/mTOR phosphorylation leading to NF-kappaB dependent AP-1-mediated ICAM-1 expression in breast cancer cells. Mol. Cancer 2010, 9, 101. [Google Scholar] [CrossRef] [Green Version]
- Chiu, T.J.; Lu, H.I.; Chen, C.H.; Huang, W.T.; Wang, Y.M.; Lin, W.C.; Li, S.H. Osteopontin Expression is Associated with the Poor Prognosis in Patients with Locally Advanced Esophageal Squamous Cell Carcinoma Receiving Preoperative Chemoradiotherapy. BioMed Res. Int. 2018, 2018, 9098215. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.J.; Zhu, Y.; Xie, K.L.; Peng, Y.P.; Tao, J.Q.; Tang, J.; Li, Z.; Xu, Z.K.; Dai, C.C.; Qian, Z.Y.; et al. Yin Yang-1 suppresses invasion and metastasis of pancreatic ductal adenocarcinoma by downregulating MMP10 in a MUC4/ErbB2/p38/MEF2C-dependent mechanism. Mol. Cancer 2014, 13, 130. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Qin, Y.R.; Bi, J.; Guo, A.; Fu, L.; Guan, X.Y. Overexpression of matrix metalloproteinase 10 is associated with poor survival in patients with early stage of esophageal squamous cell carcinoma. Dis. Esophagus 2012, 25, 656–663. [Google Scholar] [CrossRef]
- Leeman, M.F.; Curran, S.; Murray, G.I. The structure, regulation, and function of human matrix metalloproteinase-13. Crit. Rev. Biochem. Mol. Biol. 2002, 37, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Mendonsa, A.M.; VanSaun, M.N.; Ustione, A.; Piston, D.W.; Fingleton, B.M.; Gorden, D.L. Host and tumor derived MMP13 regulate extravasation and establishment of colorectal metastases in the liver. Mol. Cancer 2015, 14, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Gong, H.; Wang, J.; Tao, L.; Xu, D.; Bao, E.; Liu, Z.; Qiu, J. SENP2 regulates MMP13 expression in a bladder cancer cell line through SUMOylation of TBL1/TBLR1. Sci. Rep. 2015, 5, 13996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, N.; Ahonen, M.; Kahari, V.M. Matrix metalloproteinases in tumor invasion. Cell. Mol. Life Sci. 2000, 57, 5–15. [Google Scholar] [CrossRef]
- Martin, M.D.; Matrisian, L.M. The other side of MMPs: Protective roles in tumor progression. Cancer Metastasis Rev. 2007, 26, 717–724. [Google Scholar] [CrossRef]
- Rizki, A.; Weaver, V.M.; Lee, S.Y.; Rozenberg, G.I.; Chin, K.; Myers, C.A.; Bascom, J.L.; Mott, J.D.; Semeiks, J.R.; Grate, L.R.; et al. A human breast cell model of preinvasive to invasive transition. Cancer Res. 2008, 68, 1378–1387. [Google Scholar] [CrossRef] [Green Version]
- Vargas, A.C.; McCart Reed, A.E.; Waddell, N.; Lane, A.; Reid, L.E.; Smart, C.E.; Cocciardi, S.; da Silva, L.; Song, S.; Chenevix-Trench, G.; et al. Gene expression profiling of tumour epithelial and stromal compartments during breast cancer progression. Breast Cancer Res. Treat. 2012, 135, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, X.; Liang, Y.; Diao, X.; Chen, Q. S100A4 promotes invasion and angiogenesis in breast cancer MDA-MB-231 cells by upregulating matrix metalloproteinase-13. Acta Biochim. Pol. 2012, 59, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, H.; Liu, Z.; Yang, L.; Zhou, J.; Cao, X.; Sun, H. Circ-SLC7A5, a potential prognostic circulating biomarker for detection of ESCC. Cancer Genet. 2020, 240, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Hao, J.J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Okuno, Y.; Varela, A.M.; Ding, L.W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473. [Google Scholar] [CrossRef]
- Shi, X.J.; Chen, Z.L.; Hu, X.D.; Luo, M.; Sun, Z.M.; Li, J.G.; Shi, S.S.; Feng, X.L.; Zhou, C.C.; Li, Z.T.; et al. AJUBA promotes the migration and invasion of esophageal squamous cell carcinoma cells through upregulation of MMP10 and MMP13 expression. Oncotarget 2016, 7, 36407–36418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Shen, J.X.; Wu, H.T.; Li, X.L.; Wen, X.F.; Du, C.W.; Zhang, G.J. Collagen 1A1 (COL1A1) Promotes Metastasis of Breast Cancer and is a Potential Therapeutic Target. Discov. Med. 2018, 25, 211–223. [Google Scholar] [PubMed]
- Li, J.; Ding, Y.; Li, A. Identification of COL1A1 and COL1A2 as candidate prognostic factors in gastric cancer. World J. Surg. Oncol. 2016, 14, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oleksiewicz, U.; Liloglou, T.; Tasopoulou, K.M.; Daskoulidou, N.; Gosney, J.R.; Field, J.K.; Xinarianos, G. COL1A1, PRPF40A, and UCP2 correlate with hypoxia markers in non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2017, 143, 1133–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.X.; Lee, B.; Jiang, Y. Cell-ECM Interactions in Tumor Invasion. Adv. Exp. Med. Biol. 2016, 936, 73–91. [Google Scholar] [CrossRef]
- Yin, Y.W.; Du, L.; Li, X.Z.; Zhang, X.Y.; Gao, Y.L. miR-133a-3p suppresses cell proliferation, migration, and invasion and promotes apoptosis in esophageal squamous cell carcinoma. J. Cell. Physiol. 2019, 234, 12757–12770. [Google Scholar] [CrossRef]
- Syrjanen, K.; Pyrhonen, S.; Aukee, S.; Koskela, E. Squamous-Cell Papilloma of the Esophagus—A Tumor Probably Caused by Human Papilloma-Virus (HPV). Diagn. Histopathol. 1982, 5, 291–296. [Google Scholar]
- Syrjanen, K.J. Histological-Changes Identical to Those of Condylomatous Lesions Found in Esophageal Squamous-Cell Carcinomas. Arch. Geschwulstforsc 1982, 52, 283–292. [Google Scholar]
- Awerkiew, S.; Bollschweiler, E.; Metzger, R.; Schneider, P.M.; Holscher, A.H.; Pfister, H. Esophageal cancer in Germany is associated with Epstein-Barr-virus but not with papillomaviruses. Med. Microbiol. Immunol. 2003, 192, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.V. Human papillomaviruses and anogenital cancers. N. Engl. J. Med. 1997, 337, 1386–1388. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Liu, D.; Huang, Y.; Jiang, Z.; Wu, F.; Cen, Y.; Ma, L. Identification of Key Genes Related to the Prognosis of Esophageal Squamous Cell Carcinoma Based on Chip Re-Annotation. Appl. Sci. 2021, 11, 3229. https://doi.org/10.3390/app11073229
Wang M, Liu D, Huang Y, Jiang Z, Wu F, Cen Y, Ma L. Identification of Key Genes Related to the Prognosis of Esophageal Squamous Cell Carcinoma Based on Chip Re-Annotation. Applied Sciences. 2021; 11(7):3229. https://doi.org/10.3390/app11073229
Chicago/Turabian StyleWang, Meiqi, Dan Liu, Yunchuanxiang Huang, Ziyi Jiang, Feng Wu, Yu Cen, and Lan Ma. 2021. "Identification of Key Genes Related to the Prognosis of Esophageal Squamous Cell Carcinoma Based on Chip Re-Annotation" Applied Sciences 11, no. 7: 3229. https://doi.org/10.3390/app11073229