1. Introduction
Since the in vitro method of monoclonal antibodies (mAbs) isolation by the hybridoma technology was proposed in 1975 [
1], mAbs have become a hot topic in drug research today. Bamlanivimab and Etesevimab, which can be used to detect and treat coronavirus disease 2019 (COVID-19), have played an enormous role in controlling the pandemic [
2]. Multiple disulfide bonds combine two heavy-chain and two light-chain proteins to form a mAb with a molecular weight of about 150 kDa. The heavy and light chain’s complementary-determining region (CDR) is responsible for recognizing and binding a specific amino acid sequence or protein conformation. The considerable molecular weight of mAbs has caused some limitations in their further application, such as their poor performance in identifying hapten antigens [
3], which also makes it challenging to penetrate tissues or solid tumors [
4]. The induced anti-drug antibodies against therapeutic mAbs hindered the efficacy of clinical treatment. Moreover, the slow metabolism of labeled mAbs generated severe background signals in immunoimaging [
4]. In order to solve the problems above, partial mAb, like the antigen-binding fragment (Fab, 55 kDa) and single-chain variable fragment (scFv, ~25 kDa), are recombinantly produced. However, the affinity of those non-natural and incomplete mAb fragments is lower than the complete antibody. Furthermore, the cost of production is also higher than conventional antibodies because of the unstable molecular structure [
5,
6].
In 1993, the Hamers-Casterman team found the heavy-chain-only antibodies (HCAbs) in camelid animals (camelids and alpacas) which lack the light-chain and the heavy-chain constant region 1. The variable domain of the heavy-chain-only antibody (VHH) is recombinantly expressed as the single-domain antibody or Nanobody [
7]. VHH is small (15 kDa) and has a long CDR3 that can recognize the structure of the antigen. Therefore, the VHH is often conformation-dependent when isolated from the antibody library [
8]. Nevertheless, VHH has other advantages against a conventional antibody, as it is more resistant to proteolytic enzymes, chemical denaturants, extreme pH, and temperature changes due to its natural molecular composition [
9]. Many hydrophilic amino acids and intramolecular disulfide bonds in VHH also provide excellent solubility and thermostability [
9,
10,
11]. Other HCAbs also found in cartilaginous fishes are called immunoglobulin new antigen receptors (IgNARs) [
12]. Although the molecular weight of the variable domain of new antigen receptors (VNAR) is only 12 kDa, its CDR3 is longer than VHH, and the molecule has advantageous features similar to VHH [
13,
14,
15,
16].
Various techniques for screening mAbs were also applied to isolate antigen-specific single-domain antibodies. The phage display, ribosome display, and yeast surface display were used to build the VNAR antibody library [
17,
18,
19]. Before the screening, constructing an immune antibody library or non-immune antibody libraries, such as a naïve antibody library or synthetic antibody library, is necessary. High-affinity VNAR can be extracted from the immune antibody library, which is generated from immunization against the infectious agent as the shark’s in vivo immune mechanisms mature the antibody repertoire to yield high-affinity antibodies. But the cost of rearing animals and immunization is far greater than constructing the VHH immune antibody library [
20]. Although the VNAR affinity is moderate when isolated from non-immune antibody libraries, it has still shown a broad spectrum of specificity against antigens [
21,
22,
23]. The moderate-to-low affinity can be improved by increasing the library diversity [
24] and in vitro affinity maturation, such as the target-site mutation or saturation mutation of CDR1 [
25,
26,
27]. Another aspect of improving the affinity is to increase the diversity of the non-immune antibody library. The naïve antibody libraries are antibody libraries constructed from B cells of nonimmunized sharks. The VNAR gene sequence is isolated from a large portion of white blood cells and then cloned into display platforms for screening, requiring a large quantity of animal tissues and potentially causing some ethical issues. In contrast, the starting material for building a semi-synthetic VNAR antibody library only needs the unrearranged IgNAR-genes from B cells or the VNAR antibody gene scaffold. And at least one hypervariable loop of VNAR was genetically randomized to build the diversity of a semi-synthetic library. For example, Nuttall et al. used the striped shark VNAR as the gene scaffold and randomized the CDR3 region by saturation mutation to construct the first semi-synthetic phage VNAR library. They have successfully sorted out the VNARs against AMA-1, Tom70, or Gingipain K protease and proved that these recombinantly expressed VNARs can bind with its antigen [
18,
27,
28,
29]. In addition to the CDR3 region, the VNAR contains another three hypervariable loops, designated as CDR1, hypervariable loop 2 (HV2), and hypervariable loop 4 (HV4), respectively. The HV2 region of the VNAR gene can also be randomized to generate a new antigen-binding site for screening bispecific VNAR antibodies. Zielonka et al. employed the anti-epithelial cell adhesion molecule (EpCAM) VNAR gene as the scaffold and mutated its HV2 region to construct a semi-synthetic yeast VNAR library. Thereafter, the bispecific VNARs against EpCAM and with a cluster of differentiation of 3ε (CD3ε) or human Fcγ were screened and isolated [
30].
During the screening process to identify high-affinity binders from the yeast VNAR library, a peptide tag must be fused to VNARs to aid the analysis of the display level of the VNAR library on the surface of the yeast cell. Further, in cases where no direct fluorophore-labeled antibodies against the peptide tags are available, a tag-specific primary antibody and a fluorophore-conjugated secondary antibody might be needed for fluorescence-activated cell sorting (FACS). The complexity of the analysis and possible batch-to-batch differences of antibodies may pose reproducibility challenges to the screening process. Therefore, an improved yeast surface display platform for screening nanobody immune libraries is designed to overcome these hurdles [
31]. The nanobody library fused to the N-terminus end of Aga2p and an orthogonal acyl carrier protein (ACP) linked to the C-terminus end of Aga2p. After Sfp synthase attaches fluorophore-labeled CoA derivatives to the ACP tag, the display level of the Nanobody on the yeast cell can be monitored by fluorometry. Although this Nanobody-Aga2p-ACP fusion avoided the possible steric hindrance by a peptide tag and can be easily stained in a one-step reaction, specific reagents and extra steps are still needed in the staining process. A more straightforward strategy is employed in this study, where the GFP protein replaces the ACP tag and leads to the skipping of staining [
32]. Additionally, to speed up the VNAR discovery, a work flow was taken advantage of using the animal-free semi-synthetic yeast VNAR library and the efficient selection of antigen-specific VNAR by sequential magnetic activated cell sorting (MACS) and FACS. Eventually, a comprehensive survey of the candidate binders was done by next-generation sequencing. To prove this strategy, we screened VNARs against a feline neonatal Fc receptor (fFcRn) or feline infectious peritonitis virus (FIPV) nucleocapsid protein and verified that the selected VNARs expressed on yeast could specifically bind to the antigen.
2. Materials and Methods
2.1. Strains, Plasmids, Media, and Antibodies
Saccharomyces cerevisiae yeast display strain EBY100 (ATCC MYA-4941) was cultured at 30 °C with shaking in yeast extract peptone–dextrose (YPD) medium. The yeast cells harboring surface display vector were cultivated in synthetic dextrose casamino acids (SD-CAA) medium (14.7 g/L sodium citrate dihydrate, 4.29 g/L citric acid monohydrate, 20 g/L D(+)-glucose, 6.7 g/L yeast nitrogen base without amino acids, 5 g/L Bacto™ (ThermoFisher Scientific, Waltham, MA, USA) casamino acids, 1% penicillin-streptomycin antibiotics, 100 μg/mL ampicillin, and 50 μg/mL kanamycin). The yeast cells were grown at 20 °C in synthetic galactose casamino acids (SG-CAA) medium (7.44 g/L sodium phosphate monobasic, 5.4 g/L sodium phosphate dibasic, 20 g/L D-galactose, 2 g/L D(+)-glucose, 6.7 g/L yeast nitrogen base without amino acids, 5 g/L Bacto™ casamino acids, 1% penicillin-streptomycin antibiotics, 100 μg/mL ampicillin, and 50 μg/mL kanamycin) during induction for surface display. The
Escherichia coli strain DH5α was used for cloning and propagation of plasmid DNA. The pHEL5A7-GFP plasmid was constructed based on the sequence of the pNACP plasmid [
32]. There was a modified pNACP vector in which the acyl carrier protein (ACP) was replaced with the green fluorescent protein (GFP) [
32,
33]. The HEL-5A7 [
34] or other antigen-specific VNAR sequence was cloned into the N-terminus of Aga2p with
Hind III and
Bam HI sites in the modified pNACP cloning vector. The surface-displayed VNAR fusion protein was detected by cell-based enzyme-linked immunosorbent assay (ELISA), using Myc (human c-myc proto-oncogene) epitope tag mouse monoclonal antibody (Proteintech, Rosemont, IL, USA, catalog: 66004-1-Ig) and HRP-conjugated goat anti-mouse IgG (H + L) (Proteintech, catalog: SA00001-1). The detection of yeast clones binding to lysozyme (Sigma-Aldrich, St. Louis, MO, USA, catalog: L4919) was verified by cell-based ELISA, using rabbit polyclonal anti-lysozyme antibody (Abcam, Cambridge, UK, catalog: ab391) and HRP-conjugated goat anti-rabbit IgG (H + L) (Abcam, catalog: ab205718). When biotinylated lysozyme (Genetex, Irvine, CA, USA, catalog: GTX82960-pro) or antigen binds to yeast clones, phycoerythrin (PE)-conjugated streptavidin (Genetex, catalog: GTX85910) or peridinin–chlorophyll-protein (PerCP)-conjugated streptavidin (Biolegend, San Diego, CA, USA, catalog: 405213) is used to detect the binding complex in the fluorescence-related analysis. Allophycocyanin (APC)-conjugated streptavidin (Biolegend, catalog: 405207), biotinylated fFcRn (ACROBiosystems, Newark, DE, USA, catalog: FCN-F82W3), FIPV vaccine (Felocell
® FIP, Zoetis, San Diego, CA, USA), and biotinylated mouse anti-feline coronavirus nucleocapsid mAb (Bio-Rad, Hercules, CA, USA, catalog: MCA2594B) were also employed in the MACS and FACS analysis.
2.2. Construction of Semi-Synthetic VNAR Library
The method of constructing a semi-synthetic VNAR library was modified based on the research of Shao et al. [
35]. Using HEL-5A7 gene as PCR template, sets of NNK saturation-mutated DNA cassettes were introduced into the CDR3 loop by splice-overlap polymerase chain reaction (PCR). The first round of PCR used For-1st as forward primer, with Rev12-X3CX2CX5, Rev13-X6CX1CX4, Rev16-X6CX3CX5, Rev18-X6CX4CX6, Rev20-X5CX5CX8, or Rev23-X6CX5CX10 as the reverse primer to create six various CDR3 loop lengths (12, 13, 16, 18, or 20 and 23 amino acids) with 10, 11, 14, 16, or 18 and 21 randomized residues. The primer sequences used in this study were listed in
Table 1. Fifty microliters of the first PCR components are 11.5 μL H
2O, 10 μL 5 × Q5 buffer, 10 μL 5 × Q5 enhancer, 4 μL 2.5 mM dNTP, 5 μL 1 mM forward primer, 5 μL 1 mM reverse primer, 4 μL 2.5 ng/μL HEL-5A7 DNA template, and 0.5 μL 2 U/μL Q5 DNA polymerase (New England Biolabs, Ipswich, MA, USA). The amplification was carried out with an initial denaturing step of 98 °C for 30 s, 35 cycles of 98 °C for 5 s, 60 °C for 15 s, and 72 °C for 15 s. The second PCR with the first PCR product as a template to create 40 bp flanks was compatible with the yeast display vector, pHEL5A7-GFP, allowing cloning by yeast homologous recombination [
36]. Fifty microliters of the second PCR components are 11.5 μL H
2O, 10 μL 5 × Q5 buffer, 10 μL 5 × Q5 enhancer, 4 μL 2.5 mM dNTP, 5 μL 1 mM forward primer (For-TU1mod-40fl), 5 μL 1 mM reverse primer (Rev-TU58mod-40fl), 4 μL 25 ng/μL purified first PCR product, and 0.5 μL 2 U/μL Q5 DNA polymerase (New England Biolabs). The amplification was carried out with an initial denaturing step of 98 °C for 30 s, 35 cycles of 98 °C for 5 s, 68 °C for 15 s, and 72 °C for 15 s. The primers used in PCR were synthesized by Sangon Biotech, Shanghai, China. The primer sequences are shown in
Table 1, and the PCR products were purified with QIAquick PCR purification kit (Qiagen) before proceeding to the next step. The pHEL5A7-GFP plasmid was digested with
Hind III and
Bam HI restriction enzymes before transformation.
A yeast surface display library was generated by electroporation-mediated transformation of the second PCR product and the digested pHEL5A7-GFP vector into EBY100 yeast using the lithium acetate method [
37]. The method of electroporation-mediated transformation was modified based on the protocol by Grzeschik et al. [
38]. Each electroporation was performed using a MicroPulser electroporator (Bio-Rad), where 3 μg of PCR product and 1 μg of digested vector were added to 400 μL of electrocompetent EBY100 yeast. The electroporation was performed at 2.5 kV, 25 μF, and 200 Ω. After twelve electroporation rounds were finished, 10 mL of yeast recovery medium (1:1 mixed 1 M sorbitol and YPD medium) was added to each batch of electroporated yeast cells and incubated at 30 °C with shaking for 1 h. As the control, an electroporation reaction containing only the digested or undigested pHEL5A7-GFP vector was also performed. After incubation, the yeast cells in the recovery medium were recovered, and cells were resuspended in 1 L of SD-CAA medium. The yeast cells in SD-CAA medium were cultured at 30 °C with shaking for 36 h. Following cultivation, cells were cryopreserved and stored at −80 °C. Each cryopreservation tube contained about 1.1 × 10
10 cells.
2.3. Validation of Yeast-Surface-Displayed VNAR-GFP Fusion Protein
EBY100 yeast harboring pHEL5A7-GFP plasmid was induced in SG-CAA medium for 24, 48, or 72 h. After harvesting and washing the cells, samples were evaluated using a fluorescent microscope (Olympus® BX40F-3, Olympus, Tokyo, Japan) under the FITC filter set and bright field. The quantification of GFP fluorescence was calculated by adding 1 × 107 of induced yeast cells into a 96-well microwells plate (Corning, Corning, NY, USA, catalog: 3916), then measuring the GFP fluorescence intensity by a fluorescent ELISA reader (SpectraMax Gemini XPS/EM, Molecular Devices, San Jose, CA, USA). The GFP-positive population of induced yeast cells (~1 × 104) was estimated by a BD FACS Aria II machine (BD Biosciences, Franklin Lakes, NJ, USA) and BD FACSDiva software ver. 8.0 (BD Biosciences).
Cell-based ELISA was used to confirm that the VNAR-GFP fusion protein is indeed expressed on the yeast surface. To this end, 2 × 107 induced yeast cells were washed with PBS once and incubated at room temperature for 1 h with 100 µL of the respective Myc tag mouse monoclonal antibody, which was applied in a dilution range of 100- down to 5000-fold dilution series (1% BSA in PBS). After washing three times using PBS, 100 µL of a 1:800 dilution of HRP-conjugated goat anti-mouse IgG (H + L) in PBS with 1% BSA was applied and incubated at room temperature for 1 h. Subsequently, the cells were washed three times and incubated with 100 µL stabilized chromogen (Thermofisher, Waltham, MA, USA, catalog: SB01). The reaction was stopped by adding 100 µL of stop solution (Thermofisher, catalog: SS03100). The absorbance of cell-free supernatant was measured at 450 nm by BioTek Epoch ELISA reader (BioTek Instruments, Inc., Winooski, VT, USA).
2.4. Binding Assays for Yeast Clones against Lysozyme
In the cell-based ELISA assay, 2 × 107 induced yeast cells were washed with PBS once and incubated at room temperature for 1 h with 100 µL of lysozyme (1% BSA in PBS). After washing three times using PBS, 100 µL of a 1:5000 dilution of rabbit polyclonal anti-lysozyme antibody in PBS with 1% BSA was applied and incubated at room temperature for 1 h. Following three PBS washing steps, 100 µL of a 1:20,000 dilution of HRP-conjugated goat anti-rabbit IgG (H + L) in PBS with 1% BSA was applied and incubated for 1 h at room temperature. Subsequently, the cells were washed three times and incubated with 100 µL stabilized chromogen (Thermofisher, catalog: SB01). The reaction was stopped by adding 100 µL of stop solution (Thermofisher, catalog: SS03100). The absorbance of cell-free supernatant was measured at 450 nm by BioTek Epoch ELISA reader (Bio-Tek Instruments, Inc., USA).
In the cell-based fluorescent assay, 2 × 107 induced yeast cells were washed with PBS once and incubated at room temperature for 1 h with 100 µL of biotinylated lysozyme (1% BSA in PBS). After washing three times using PBS, 100 µL of a 1:100 dilution of PE-conjugated streptavidin in PBS with 1% BSA was applied and incubated at room temperature for 1 h. Subsequently, the cells were washed three times and resuspended with PBS. These cells were added into a 96-well microwells plate (Corning, catalog: 3916), then the GFP and PE fluorescence intensity were measured by a fluorescent ELISA reader (SpectraMax Gemini XPS/EM, Molecular Devices, San Jose, CA, USA). The PE to GFP ratio is calculated by dividing PE readings by GFP readings.
In the flow cytometry binding assay, 5 × 107 induced yeast cells were washed with PBS once and incubated with biotinylated lysozyme (1% BSA in PBS) at room temperature for 1 h. After washing three times using PBS, 100 µL of a 1:100 dilution of PE-conjugated streptavidin in PBS with 1% BSA was applied and incubated at room temperature for 1 h. The cells were washed three times and resuspended with PBS for subsequent flow cytometry analysis by Moflo™ XDP high-performance cell sorter (Beckman Coulter, Brea, CA, USA). The data were analyzed using Summit software ver. 5.3 (Summit Software, Little Rock, AR, USA). At least 3.5 × 105 yeast cells per sample were analyzed and grouped based on discrimination of GFP and PE fluorescence intensity, representing those yeast cells displaying VNAR on the surface and binding biotinylated lysozyme.
2.5. Screening Feline Neonatal Fc Receptor-Specific VNAR Displaying Yeast Clones by Sequential MACS and FACS
2.5.1. Magnetic-Activated Cell Sorting
The negative selection against magnetic beads was performed to selectively remove streptavidin and bead-specific clones. Briefly, 5 × 109 induced semi-synthetic VNAR library cells were incubated with 4 × 107 magnetic beads (Dynabeads™ Biotin Binder, Invitrogen, Waltham, MA, USA, catalog: 11047) at 4 °C for 2 h. After removing the magnetic beads by a DynaMag magnet (DynaMag™-Spin Magnet, Invitrogen, catalog: 12320D), new magnetic beads were added to start the next round of negative selection.
After three rounds of negative selection, one round of positive MACS selection against fFcRn was conducted before FACS. To prepare the fFcRn-labeled magnetic beads, 4 × 107 magnetic beads were incubated at 4 °C for 2 h with 33 pM biotinylated fFcRn (ACROBiosystems, catalog: FCN-F82W3) in PBS with 1% BSA. Magnetic beads were collected by a DynaMag magnet and washed with PBS twice. Finally, the magnetic beads were resuspended in PBS of pH 6 with 1% BSA and stored at 4 °C. After the negative MACS selection, yeast-displayed VNAR library cells were incubated with 4 × 107 fFcRn-labeled magnetic beads at 4 °C for 2 h (1% BSA in PBS of pH 6). The yeast bound magnetic beads were washed three times and resuspended with SD-CAA medium. The MACS-sorted yeasts were cultured at 30 °C with shaking in SD-CAA medium. The enriched MACS-sorted yeasts were induced in SG-CAA medium at 20 °C for 72 h. After induction, the cells were washed with PBS once and resuspended in PBS of pH 6 with 1% BSA and stored at 4 °C.
2.5.2. Fluorescence-Activated Cell Sorting
After a round of positive MACS selection, 5 × 107 induced VNAR library cells were incubated at 4 °C for 1 h with or without 500 nM biotinylated fFcRn in PBS of pH 6. To monitor the enrichment of unwanted clones during sorting, cells not incubated with the antigen were used as the control group. These control cells were incubated with fluorescent-conjugated streptavidin but without the addition of the biotinylated antigen. The cells were washed with PBS once and incubated at 4 °C for 1 h with 100 μL of a 1:100 dilution of APC-conjugated streptavidin in PBS of pH 6. After the cells were washed with PBS once and resuspended in PBS of pH 6 with 1% BSA, a two-color flow sort was performed through a MoFlo XDP Flow Cytometry Sorter. The data were analyzed using Summit software ver. 5.3. The sorted cells were grown in SD-CAA medium and induced in SG-CAA medium before the next round of selection. The concentration of the biotinylated fFcRn used was 500 nM in each round of sorting. Control experiments in which the library yeasts were incubated with the APC-conjugated streptavidin alone were performed to evaluate the undesired enrichment of clones that bind to the fluorescent dye. Plasmids from an aliquot of post-sorted yeast cells were extracted utilizing the Zymoprep Yeast Plasmid Miniprep kit I (Zymoresearch, Irvine, CA, USA, catalog: D2001) for further identification of the clone’s sequence.
2.6. Screening Feline Infectious Peritonitis Virus Nucleocapsid-Binding VNAR Displaying Yeast Clones by Sequential MACS and FACS
2.6.1. Magnetic-Activated Cell Sorting
The negative selection against magnetic beads was performed to selectively remove mouse mAb, streptavidin and bead-specific clones. Firstly, 4 × 107 magnetic beads were incubated at 4 °C for 2 h with 6 μL of biotinylated mouse anti-feline coronavirus monoclonal antibody (Bio-Rad, catalog: MCA2594B) in 1 mL PBS with 1% BSA. Magnetic beads were collected by a DynaMag magnet and washed with PBS twice. The magnetic beads were resuspended in 5 × 109 induced semi-synthetic VNAR library cells and incubated at 4 °C for 2 h. After removing the magnetic beads with a DynaMag magnet, new mouse anti-feline coronavirus mAb-labeled magnetic beads were added to start the next round of negative selection.
After three rounds of negative selection, one round of positive MACS selection against FIPV nucleocapsid was conducted before FACS. To prepare the FIPV nucleocapsid-labeled magnetic beads, 4 × 107 mouse anti-feline coronavirus mAb-labeled magnetic beads were incubated at 4 °C for 2 h with 1/10 of the reconstituted FIPV vaccine (Felocell® FIP, Zoetis) in PBS with 1% BSA. Magnetic beads were collected by a DynaMag magnet and washed with PBS twice. Finally, the magnetic beads were resuspended in PBS with 1% BSA and stored at 4 °C.
After the negative MACS selection, the yeast-displayed VNAR library cells were incubated with 4 × 107 FIPV nucleocapsid-labeled magnetic beads at 4 °C for 2 h (1% BSA in PBS). The yeast-bound magnetic beads were washed three times and resuspended with SD-CAA medium. The MACS-sorted yeasts were cultured at 30 °C with shaking in SD-CAA medium. The enriched MACS-sorted yeasts were induced in SG-CAA medium at 20 °C for 72 h. After induction, the cells were washed with PBS once and resuspended in PBS with 1% BSA and stored at 4 °C.
2.6.2. Fluorescence-Activated Cell Sorting
After a round of positive MACS selection, 5 × 107 induced VNAR antibody library cells were incubated at 4 °C for 1 h with 1/3 of the reconstituted FIPV vaccine in PBS with 1% BSA. The cells were washed with PBS once and incubated at 4 °C for 1 h with 100 μL of a 1:100 dilution of biotinylated mouse anti-feline coronavirus mAb in PBS with 1% BSA. After, the cells were washed and 100 μL of a 1:100 dilution of APC-conjugated streptavidin in PBS with 1% BSA was added and incubated at 4 °C for 1 h. Finally, the cells were washed with PBS once and resuspended in PBS with 1% BSA. A two-color flow sort was performed through a MoFlo XDP Flow Cytometry Sorter. The data were analyzed using Summit software ver. 5.3. The sorted cells were grown in SD-CAA medium and induced in SG-CAA medium before the next round of selection. The ratio of the reconstituted FIPV vaccine used was 1/3 in each round of sorting. Control experiments in which the library yeasts were incubated with the biotinylated mouse anti-feline coronavirus mAb alone were performed to evaluate the undesired enrichment of clones that bind to the mAb. Plasmids from an aliquot of post-sorted yeast cells were extracted using the Zymoprep Yeast Plasmid Miniprep kit I to identify the clone’s sequence.
2.7. Binding Assay for FACS-Sorted Yeast Clones against Antigen
The cell-based fluorescent assay was employed to verify the binding of fFcRn-specific yeast clones to fFcRn and the binding of FIPV nucleocapsid-specific yeast clones to FIPV nucleocapsid. VNAR sequences from post-sorted sequences analysis were picked and subcloned into the modified pNACP vector via Hind III and Bam HI sites. These constructed plasmids were transformed into EBY100 yeast. VNAR-displaying yeast clones were inoculated and induced for the binding assay.
For the analysis of fFcRn-specific yeast clones, 2 × 107 induced VNAR-displaying yeast cells were washed with PBS once and incubated at 4 °C for 1 h with 250 nM biotinylated fFcRn or biotinylated lysozyme in PBS of pH 6 with 1% BSA. The cells were washed with PBS once and incubated at 4 °C for 1 h with 100 μL of a 1:100 dilution of PerCP-conjugated streptavidin in PBS of pH 6. Subsequently, the cells were washed three times and resuspended with PBS. These cells were added into a 96-well microwells plate, then the GFP and PerCP fluorescence intensity were measured by a SpectraMax Gemini XPS/EM reader. The PerCP to GFP ratio is calculated by dividing PerCP readings by GFP readings.
For the analysis of FIPV nucleocapsid-specific yeast clones, 2 × 107 induced VNAR-displaying yeast cells were washed with PBS once and incubated at 4 °C for 1 h with 1/10 of the reconstituted FIPV vaccine or 250 nM biotinylated lysozyme in PBS with 1% BSA. The cells were washed with PBS once and incubated at 4 °C for 1 h with 100 μL of a 1:100 dilution of biotinylated mouse anti-feline coronavirus monoclonal antibody in PBS with 1% BSA. After, the cells were washed and 100 μL of a 1:100 dilution of PerCP-conjugated streptavidin in PBS with 1% BSA was added and incubated at 4 °C for 1 h. Subsequently, the cells were washed three times and resuspended with PBS. These cells were added into a 96-well microwells plate, then the GFP and PerCP fluorescence intensity were measured by a SpectraMax Gemini XPS/EM reader. The PerCP to GFP ratio is calculated by dividing PerCP readings by GFP readings.
2.8. The Calculation of Normalization and Fold Enrichment after FACS Sorting
The fraction of all GFP-positive cells and GFP
+APC
+ cells can be obtained in each round of FACS screening. The ratio of observed double-positive cells was normalized to all GFP-expressing cells. The normalization is calculated using the following equation:
The FACS enrichment factor is calculated from the normalized frequency of GFP
+APC
+ cells after the FACS sort divided by the normalized frequency of the pre-sorted cells using the following equation:
2.9. Next-Generation Sequencing
Next-generation sequencing was performed on the input library and every round of sorted samples using the Illumina MiSeq platform on VNAR CDR3 region amplified from the yeast plasmids. Plasmids from an aliquot of pre- and post-sorted yeast cells were used as the PCR template, using primers NGS-F (5′-GAAACGAGGAGAGCATATCG-3′) and NGS-F (5′-ATCCATTCACAGTCACGG-3′) to amplify the gene fragment containing VNAR CDR3. PCR reactions to add short universal adaptor sequences on the 5′ and 3′ ends and add barcoded adaptor sequences were accomplished by Topgen Biotechnology (Kaohsiung City, Taiwan). Amplicons were sequenced on an Illumina MiSeq system using 2 × 250-bp paired-end reads. Acquired NGS data were also processed by Topgen Biotechnology. Briefly, Trim Galore! (v0.4.3.1) was used to check the quality of the original data and remove the barcode sequence, and then the paired-end sequences were merged into a single sequence by Fastq-join (v1.1.2-806). FastQC (v0.72) and MultiQC (v1.8) were employed to ensure that the PHRED scores are greater than 30 and that the sequence length and type must be within the preset gene mutation scaffold. The amplification factor was calculated from the frequency of a sequence before FACS sorting divided by the frequency of the sequence after sorting. The amino acid frequency was calculated using Microsoft Excel scripts. The identification of consensus amino acid sequence before and after FACS sorting was based on the statistical significance of the individual residues in contrast to a background frequency, and was calculated and visually presented by the pLogo [
39].
2.10. Statistical Analysis
Student’s t-test and one-way analysis of variance (ANOVA) was used to analyze the data. When ANOVA identified differences among the groups, Duncan’s multiple range test was conducted to examine significant differences among the treatments in SAS software 9.4 (SAS Institute, Cary, NC, USA). The level of significance was p < 0.05.
4. Discussion
We developed a rapid and improved platform for screening affinity VNARs by MACS and FACS, followed by next-generation sequencing analysis. By the property of GFP, our system allows us to monitor the VNAR display level on each yeast clone. We validated this platform by selecting affinity VNARs against purified and non-purified proteins. The entire process does not involve animal immunization, which is the crucial step of antibody isolation. Generally, the immunization process for preparing immune antibody libraries takes 2 to 4 months. Milligrams of highly purified antigens are required for immunization [
8], and it is necessary to consider whether the antigen is immunogenic or toxic to animals. The in vitro antibody discovery process can strictly control the screening environment, such as screening antibodies under different pH environments [
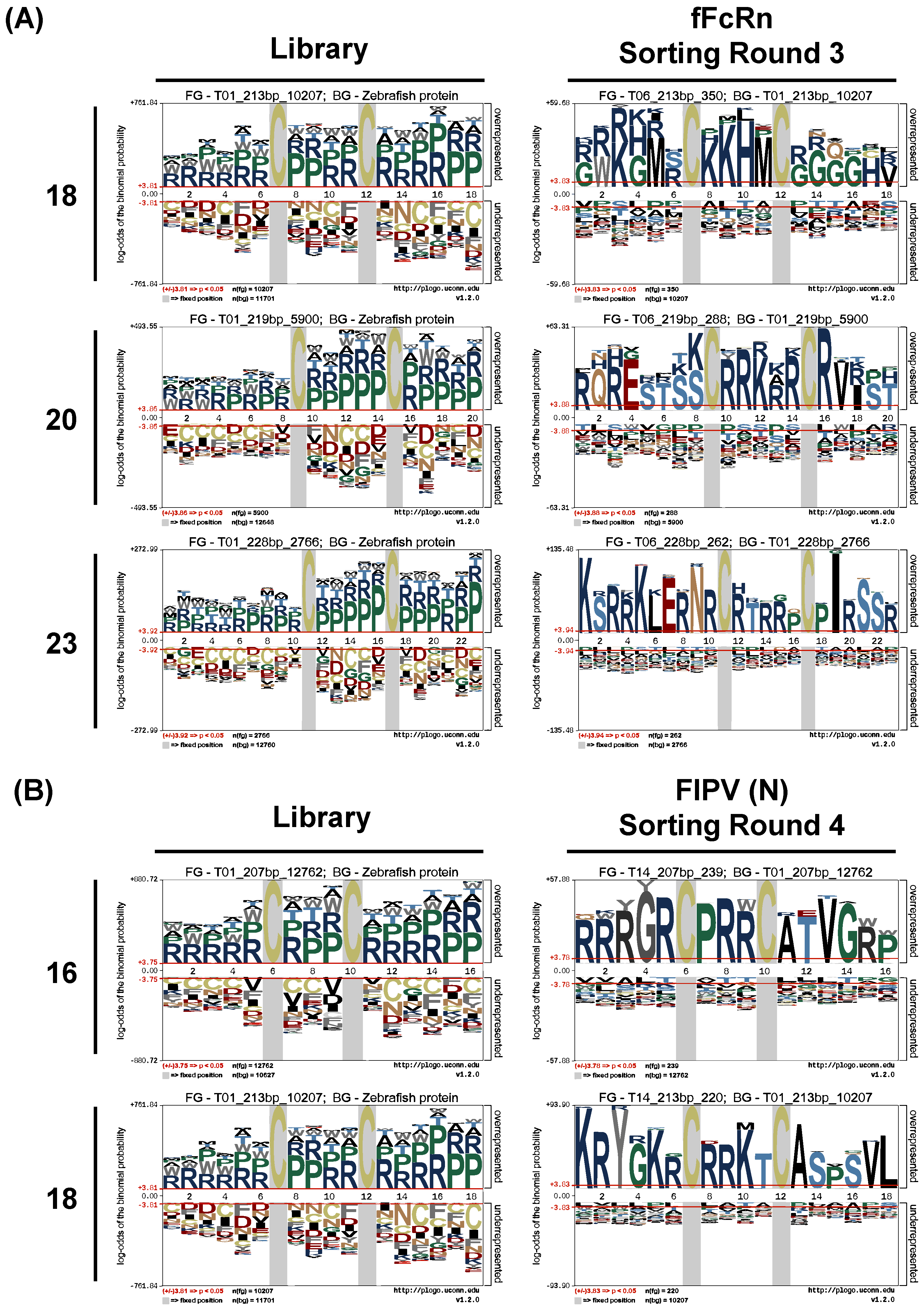
47] or screening for antibodies that can bind to different conformations of antigens without re-immunization. The yeast clones evaluated by NGS analysis can avoid losing potential VNAR candidates. The frequency and amplification factor analysis based on NGS data can also help exclude many non-target clones in the last screening stage to accurately select the functional VNAR sequence. The pLogo analysis can be used to extract consensus antigen-binding CDR3 motifs which may be helpful to design the in vitro affinity maturation experiment.
The yeast agglutinin display expression system can be divided into α-agglutinin and a-agglutinin display expression systems. The C-terminus of the anchoring protein in an α-agglutinin display expression system is used for connecting to the yeast surface [
48]. Therefore, only the N-terminus of the anchoring protein is available for expressing the heterologous protein. In contrast, the anchoring protein Aga2p of the a-agglutinin display expression system uses disulfide bonds to connect with the yeast surface, so the N-terminus and C-terminus of Aga2p are unrestricted to express heterologous proteins [
49]. Thus, a dual display of proteins on the yeast cell surface was established to quantify binding reactions or enzymatic bioconjugation reactions between both ends of Aga2p [
49]. In this study, the GFP protein was expressed at the C-terminus of Aga2p, and the VNAR antibody library was placed at the N-terminus, since the orientation of protein expression in a eukaryotic cell is from the N-terminus to the C-terminus. If the GFP protein is functionally expressed, the VNAR antibody is also well expressed. Moreover, the cell-based ELISA results prove that this study’s co-expressed GFP and VNAR are functional. Generally, before the FACS sorting of yeast, it is necessary to fluorescently specify the surface-expressed protein and the antigen bound to it. In this dual display system, an anchored VNAR fusion protein can emit GFP fluorescence spontaneously and then label the bound antigen for screening. After that, the dual fluorescence can be detected by a fluorescent ELISA reader or analyzed and sorted by a flow cytometer.
The VNAR antibody library established in this study retains two conserved cysteines and four atypical cysteines in the HEL-5A7 gene scaffold. Other than the variation of the cysteine number, the two-cysteine CX
nC motifs in the CDR3 region are discovered in the variable domain of the antibody heavy chain by NGS analysis [
50]. This mega research can be a start point to design a new antibody with long CDR3 or cross-reactive activity. Some studies of synthetic antibody libraries choose to remove cysteines to reduce the formation of disulfide bonds because it will limit the choice of subsequent antibody expression systems [
38,
51]. The study of Li et al. mutated the non-canonical cysteines of CDR1 and CDR3, because they want to improve the likeness to the variable domain of human heavy chains [
52]. However, disulfide bonds can form tighter protein structures, increasing protein stability and assisting the finger-like structure formed by CDR3 in recognizing antigenic epitopes deep in protein clefts [
53]. Another study also shows that the number of cysteines in the synthetic VNAR antibody library does not affect the screening efficiency, which means that the high-affinity binders can be screened and produced [
54].
The diversity of the antibody library is the key to screening high-affinity binders. To yield nM affinity binders, the library capacity must be higher than 10
9 [
55]. But the diversity of a typical yeast-derived library does not exceed 10
7, becoming the bottleneck of the isolating high-affinity binder. In this study, the method of maximizing the diversity of the VNAR library is based on Shao et al.’s research on the semi-synthetic VNAR phage display library, which contains greater than 1 × 10
8 unique phage clones [
35]. An entirely random NNK trinucleotide combination was used to create different lengths of site mutations in the CDR3 region by PCR, and the theoretical diversity can be as high as 10
14~10
27 in this study. Although the theoretical diversity is high, many factors will affect the actual diversity. For example, the efficient transformation of foreign DNA into yeast could be a limitation. The library bias created by the NNK mutagenesis also affects the expected diversity with the distorted amino acid frequency. The NNK codon covers 32 codons that include all amino acids, but with redundancy for 8 amino acids (Arg, Ser, Leu, Pro, Ala, Thr, Gly, and Val). The most abundant amino acids observed in our library mostly fit the expected frequency based on the NNK codon which means that the frequency bias does exist in our study (
Figure 4). The frequency bias also has impact on the pLogo analysis. The two most frequent amino acids, arginine and proline, occupied almost all the positions of randomized CDR3 before sorting (
Figure 4). This phenomenon may hinder the finding of the high-affinity binder because the translation-based bias might force the antigen binds to the less suitable clone. Each NNK mutation site also has a chance of mutating into a stop codon (3%), undesired cysteine (3%), or hydrophobic residues (27%), which may cause the low efficiency of the VNAR display. Therefore, the longer the CDR3 is, the higher the chance of a difficult translation. In our study, for the CDR3 with lengths of 12, 13, 16, 18, 20, or 23 residues, the proportion of complete VNAR sequences decreased with the CDR3 length, suggesting that another translational bias happened in our library. Nevertheless, the actual diversity in this study could still maintain a level of more than 10
9 after checking the library quality by the NGS assay. Even though the library’s capacity is larger than usual, the bio-panning efficiency is low when the input library overreaches the FACS workload [
56]. Hence, we employed the strategy of sequential MACS and FACS to process a large quantity of library yeast. At first, the MACS filtered out possible binders from the whole library regardless of the low sorting purity. Next, these MACS-sorted clones, which contained true-positive or false-positive clones, were purified by FACS. Our data show that after the final round of fFcRn FACS sorting, the frequency of GFP
+APC
+ cells in the control group increased. A similar result was found in the isolation of FIPV nucleocapsid-specific VNARs, showing that the enrichment of off-target clones is designed to happen in the end. To avoid this phenomenon, a more rigid negative selection during MACS or changing fluorescent dye between rounds of panning could be the solution. However, it means more delays and costs in the development process. We thought to speculate that if the NGS assay can be applied to compare the sorted sequences in every round of the control group, the unwanted sequences can be excluded from sorted candidates easily without needing more work and changing reagents.
Biopanning is a standard process for screening affinity clones from the phage antibody library. In each round of screening, the bound phage must be stripped from the antigen-coated ELISA plate. Many strongly bound phages cannot be detached from the antigen and disappear in the next round of screening. In addition, after multiple rounds of biopanning, the screened clones can be dominated by phages with moderate affinity or rapid replication. Usually, 10
4~6 phages were panned out, but only 100~1000 phages will be hand-selected for sequencing, and the opportunity to find high-affinity clones is diminished [
41]. Although biopanning by FACS does not lose the strong bound clones during sorting, the sorted populations may still be occupied by the fast-replication clones. In this study, all sorted yeasts were sequenced by NGS, so the influence of fast-replication clones could be reduced. The study of Deschaght et al. combined two analysis methods (sequence frequency > 10 and amplification factor > 10) to narrow the 2.7 × 10
5 VHH phage clones down to 5173 clones. In total, 28 clones were selected for expression and validation for antigen binding, and 25 of 28 clones were high-affinity binders. However, the affinity of VHH phage clones is not highly correlated with the sequence frequency and amplification factor [
57]. Another method to remove fast-propagating sequences or biased sequences is the bioinformatics processing tools such as the pLogo analysis to extract specific-binding sequences. In our platform, two antigens were screened for high-affinity binders, and the results of the NGS sequencing were grouped according to the sequence frequency and amplification factor analysis. Two of each most abundant and enriched antigen-specific CDR3 sequences are selected for further validation. Interestingly, these sequences are identical or highly consistent to the pLogo extracted motif (V6 sequence KSRRKLERNRC-RTRRPCPIRSSR versus 23-mer motif KSRRKLERNRCRTRRPCPIRSSR; V5 sequence RQRESTSSCRRKRRCRVRST versus 20-mer motif RQRESTSSCRRKRRCRVIST; V3 sequence KRYGKRCRRKTCASPSVL versus 18-mer motif KRYGKRCRRKTCASPSVL; and V2 sequence RRRGRCPRRCATVGRP versus 16-mer motif RRRGRCPRRCATVGWP). The frequent occurrence of arginine and proline in the CDR3 motif may be due to the influence of frequency bias. But arginine and proline can be served as protecting osmolytes to stabilize protein folding and increase protein solubility, inhibiting protein aggregation [
58]. Thus, the retained arginine and proline in the consensus CDR3 motif might be the result of a secondery selection pressure other than the target-binding. The VNAR protein structure with medium affinity may be robust compared to the high-affinity one and then stably presented on the yeast surface during biopanning. Although it seems that the result of biopanning is affected by the translation-based bias, the cell-based fluorescent assay results verified that the selected VNAR-displaying clones are able to bind to their ligand. In the end, their affinity needs to be determined after being solely expressed to verify the readiness of the grouping strategy. Such VNAR-displaying yeasts may also represent a kind of powerful point-of-care diagnostic tool for replacing current immunomagnetic beads due to the high-density, organized and robust VNAR presented on the yeast surface. We will look forward to developing a rapid FIPV detection kit in the future.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}