
Oxygen Self-Diffusion in Fluorite High Entropy Oxides
1
Department of Electrical and Computer Engineering, University of Thessaly, 38221 Volos, Greece
2
Department of Materials, Imperial College London, London SW7 2BP, UK
Appl. Sci. 2024, 14(12), 5309; https://doi.org/10.3390/app14125309
Submission received: 14 May 2024
/
Revised: 4 June 2024
/
Accepted: 17 June 2024
/
Published: 19 June 2024
(This article belongs to the Section Materials Science and Engineering)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:High-entropy oxides have recently attracted the interest of the community as a way of attuning the properties of oxides to energy applications. Here, we employ molecular dynamics simulations combined with empirical pair potential models to examine the predicted oxygen diffusivity of fluorite-structured high-entropy oxides. We show that lower levels of the dopants increase the overall diffusivity of the composition, but not to the levels of diffusion seen in yttria-doped zirconia. We attribute this to an increased resistance of the cation sublattice to the distortion that occurs through any multiple substitutions on the cation sublattice. To conclude, it is calculated that oxygen self-diffusion in high-entropy oxides is suppressed as compared to isostructural ternary oxides.
1. Introduction
There is a drive to maximize ionic diffusion for more efficient performance of the energy materials presently considered for electrochemical solid-state devices (fuel cells and battery applications) [1,2]. For example, in solid oxide fuel cells (SOFC), the operation at intermediate temperatures requires materials with lower activation energies of diffusion to compensate [3,4]. This is because, typically in SOFCs, the oxygen vacancy content and the mobility of the electrode material are linked to the surface exchange kinetics and impact the cathode performance [3,4]. Accelerating ionic diffusion in ceramics can be achieved by doping, mechanical means (strain engineering), and designing the morphology (for example, grain boundary engineering and nanocomposites) [5,6,7,8,9]. In recent studies, Rost et al. [10] introduced the High-Entropy Oxides (HEO) concept, in which mixed oxides are stabilized by entropy. HEO are in essence analogous to the High-Entropy Alloys (HEA), which are alloys where high configurational entropy (∆Sconfig) that can stabilize metastable single phases (temperature is adequately high so as to account for the negative formation enthalpies of secondary phases) typically via high temperature sintering [11,12,13].
Several studies have examined the oxygen ion self-diffusion in fluorite-structured oxides [14,15,16,17]. Although the use of high-entropy compositions greatly expands the number of potential compounds, the values of oxygen diffusion have been shown to be relatively unremarkable and, in most cases, do not exceed the values reported for archetypal ternary oxides, such as ZrO2 + 8% Y2O3 (designated Z8Y). Characteristically, in the study of fluorite HEO, Gild et al. [15] synthesized numerous single-phase fluorite oxides. These solid solutions had equal molar fractions of HfO2, ZrO2, and CeO2 as the base materials, whereas the oxides of yttrium (Y), gadolinium (Gd), ytterbium (Yb), titanium (Ti), magnesium (Mg), calcium (Ca), and lanthanum (La), in turn acted as fluorite structure stabilizers [15]. Irrespective of the composition, the determined activation energies of oxygen self-diffusion are unspectacular (1.14–1.29 eV) and the conductivity values are nearly an order of magnitude lower as compared to Z8Y [15]. The low values of ionic diffusivity are likely due to trapping of oxygen ion vacancies by the high concentrations of aliovalent cation dopants used in the construction of near-equimolar HEOs. Therefore, one strategy to improve the ionic diffusivity is to reduce the level of aliovalent doping, replacing it with mixed but isovalent dopants.
What is evident in the literature is that irrespective of a number of high-level experimental and theoretical studies (for example, Refs. [18,19,20,21,22,23,24,25,26,27] and the references therein), there is no understanding on the impact of entropy stabilization on the oxygen self-diffusion in fluorite HEO. The key question is as follows: Does it improve the oxygen self-diffusion or not? In an attempt to address this in the present study, we use molecular dynamics (MD) simulations coupled with classical interatomic potentials to study the impact of composition and, in particular, the influence of the level of trivalent rare-earth dopants upon the oxygen ion diffusivity of a series of high-entropy alloy compositions. The focus will be on the HEOs of Zr(1−x)/3Hf(1−x)/3Ce(1−x)/3Gdx/2Yx/2O2−x, Zr(1−x)/3Hf(1−x)/3Ce(1−x)/3Yx/2Lax/2O2−x at x = 0.14 (8%RE2O3) compared with predicted oxygen ion diffusivity for Z8Y and C8Y.
2. Methodology
Molecular dynamics calculations were performed using the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code [28,29]. We employed classical molecular statics and dynamic simulations using the Buckingham empirical potentials [30,31], the transferability of which has been tested in previous studies [32,33]. Static transition state atomistic simulations were performed using the GULP code [34,35]. Entropy-stabilized oxides posed challenges for the atomic scale simulation methods as the many different cations led to a great number of potential configurations, all of which may have influence over the overall bulk properties. Although classical potentials are of the order 104 times faster than the electronic structure calculations, automated methods for producing simulation data are still necessary.
For the materials considered here, the following method was adopted to provide an estimate of the mean squared displacement at an average temperature T:
- An 8 × 8 × 8 supercell of the fluorite structure is created with a random assignment of cations according to a specified stoichiometry. Oxygen ion vacancies are introduced onto the oxygen ion sublattice to provide charge balance.
- An equilibration series of simulations which consist of the following:
- Annealing is at a set temperature of 1500 K to equilibrate the distribution of oxygen vacancies.
- A 2 ns simulation at constant (zero) pressure a set temperature of Tset is used to establish the equilibrium cell parameters at temperature.
- The cell is then set to the average value of the lattice parameters, and a further 50 ps of simulation time is run to allow the vacancy distribution to adapt to the altered cell volume.
- The statistics run is calculated in a constant volume, a constant energy ensemble (i.e., without the thermostat) for a simulation time of 10 ns or the time required for the mean squared displacement of the oxygen ions to reach 10 Å2. The latter condition allows for efficient early termination of compositions with clear values of diffusivity. The simulation time and displacement of the ions R(t) is recorded in this last step and used to calculate the average temperature T (~Tset) and mean squared displacement () of the oxygen ions.
The oxygen ion diffusivity, D(T,conf.) of each configuration is then estimated by a least squares fit of the Einstein relation to the long timescale behavior of the displacement R(t) at time t as follows:
From these data, we can now characterize each composition by fitting an Arrhenius relationship to each set of T, configuration data point to obtain a value for the activation energy and preexponential for a given composition assuming the following:
3. Results and Discussion
In the present work, we studied three general compositions of high-entropy oxides based upon reported experimental synthesis and the availability of oxygen ion potentials to describe their behavior. These are Zr(1−x)/3Hf(1−x)/3Ce(1−x)/3Gdx/2Yx/2O2−x (HEO_A), Zr(1−x)/3Hf(1−x)/3Ce(1−x)/3Yx/2Lax/2O2−x (HEO_B) and Zr(1−x)/2Hf(1−x)/2Yx/3Lax/3 Prx/3O2−x (HEO_C).
Figure 1 shows the calculated values of diffusivity against temperature for the two considered HEOs compared with the calculated values for CeO2 + 8% Y2O3, C8Y, and ZrO2 + 8% Y2O3, Z8Y. At these compositions, the oxygen ion diffusivity is generally lower than the ternary oxides. We see generally good agreement with the Arrhenius behavior, and the fitted values of activation energy are in agreement with those reported in the literature. Figure 1 also shows the importance of a statistical approach to the calculations. For a given temperature, there is a scatter in the range of values. For Z8Y, this is quite small but for the HEO compositions, there are significant differences in diffusivity for different configurations within a single composition, and this is evident even with the quite large 6000 atom cells considered here.
Top image of Figure 2 shows the ionic diffusivity values at 1000 K for the three HEO compositions and ZrO2 + Y2O3 for comparison as a function of the total trivalent dopant concentration, i.e., the sum of the La, Y, Pr and Gd molar fractions. The datapoints for ZY show a rapid increase with Y-doping, a maximum around 10% molar fraction Y (~6% Y2O3), and a slow decrease in diffusivity. This behavior has been extensively described in the literature as a balance between the creation of charge compensation oxygen vacancies and the trapping of the vacancies through the addition of large densities of trivalent ions. Similar curves are observed for the HEO compositions; however, in these cases, the overall diffusivity is generally lower, particularly for the maximum peak value of diffusivity at around 10% dopant concentration. We explored this behavior in middle and bottom images of Figure 2, where the points show the predicted values of D0 and Ea for the different compositions. Middle image of Figure 2 indicates that the activation energy for diffusion for ZY decreases to a value of 0.4 eV, comparable with that of oxygen vacancy diffusion in the dilute limit. For the HEOs, however, the activation energy for diffusion tends to be a significantly higher value of around 0.6–0.75 eV, depending upon the composition. The value of D0 calculated from the simulations varies with total aliovalent doping concentration but is relatively unchanged between the different compositions considered. The decrease in the diffusivity in the HEOs as compared with the ZY is therefore due to this increased activation energy, particularly when comparing the data up to ~10% molar dopant concentration.
We considered why the HEO compositions exhibited these levels of oxygen ion diffusion, focusing specifically on the reduced diffusion because of the increased activation energy exhibited in top and middle images of Figure 2. Oxygen diffusion is mediated via vacancies moving through the fluorite lattice, a process that, in the doped materials, is hindered both by vacancy–vacancy interactions, Coulomb attraction, and trapping by the trivalent dopant ions. Figure 3a shows an illustration of the fluorite lattice with an example oxygen vacancy in the nearest neighbor position to a Y3+ ion, and the minimum energy pathway for this cell as the vacancy exchanges places with an adjacent oxygen ion. Figure 3b shows both the energy maxima at the transition point and the finite reaction energy, Er, as the sites are not symmetrically equivalent.
To help us understand the effect of multiple substitutions at the cation sites, we defined the barrier energy Eb, which is a first-order approximation to the transition barrier if Er was zero, and there were no energetic difference between the two sites.
where Ets is taken as the maximum energy difference from the lower end of the transition start or end points. Physically Eb corresponds to the maximum energy required to deform the lattice that is required to accommodate the ion as it moves through the transition pathway, and ½ Er represents the trapping of the oxygen vacancies by the host cations.
Figure 4 shows plots of the averages and spread of values of Eb and ½ Er for the oxygen vacancy transition state calculations performed at random configurations of cations, with 6.25% Y and various levels of isovalent cation doping from zero Ce + Hf to a total of 66%, i.e., an equimolar amount of Zr, Hf, and Ce. These data show that the value of the activation energy increases with isovalent doping, in agreement with the low Y-doping MD simulation results shown in Figure 2. The increase in activation energy is, however, driven primarily by an increase in the trapping of oxygen vacancies by the host lattice, Er, such that the average and spread of the Eb values remain relatively unchanged with increasing amount of cation mixing.
Here, we considered MD calculations to determine if the formation of fluorites HEOs have better self-diffusion properties as compared to more conventional fluorite-structured materials. In the literature, there is substantial experimental work on HEO; nevertheless, there is a lack of systematic understanding on oxide–ion conductivity and ion mobility as a consequence of entropy stabilization. This is further complicated when considering the extensive compositional range of fluorite HEOs. Additionally, it is established that in aliovalent-doped stabilized zirconia, the substitutional cation concentration reaches an optimum where there is a balance between the oxygen vacancy concentration (i.e., the vehicle of diffusion) with the dopant–vacancy trapping effects. Considering that this optimum doping content is nearly impossible to predict beforehand in HEOs, further work will be required.
For many years, simple interatomic potentials have proven their transferability and potential to describe diffusion properties in oxides with higher accuracy as compared to experiment. At any rate, many of the classical interatomic potentials used may not capture all the subtleties of interatomic interactions in complicated systems such as HEOs. In that respect, ab initio calculations should be used to clarify if these subtleties will lead to a better understanding of diffusion properties [36,37]. The advantage of classical simulation is the ability to perform simulations on extended systems that capture the dopant–defect interactions with respect to the local environments. Conversely, ab initio molecular dynamics (AIMD) simulations are typically limited to smaller systems (up to a few hundreds of atoms at best) and sub-nanosecond timescales. This, in turn, can result in far fewer diffusion events (particularly in high activation of diffusion systems) and poor statistics [37]. At any rate, present and near future computational resources will allow more rigorous AIMD calculations, which can enrich our understanding of these systems with electronic structure details and insights that cannot be obtained by classical MD studies. Furthermore, there is grounds for further analysis of the simulation results via the appropriate thermodynamic models that to link microscopic and macroscopic properties, such as the cBΩ model by Varotsos and Alexopoulos [38,39,40].
4. Conclusions
In this paper, we have examined the oxygen ion diffusivity as predicted from a series of molecular dynamics simulations of typical fluorite-structured high-entropy oxides. We have shown that the values of diffusivity are predicted to be generally lower than isostructural ternary oxides such as ZrO2 + Y2O3. This is reflected by the oxygen diffusion activation energy of HEO, which is more than 0.2 eV higher as compared to the ternary oxides. Therefore, the present results show that the formation of HEOs are not beneficial for oxygen self-diffusion. The present study aims to motivate further experimental work on the direct comparison between HEO and comparable fluorite compositions to conclusively determine whether HEOs provide any benefit.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Acknowledgments
The author acknowledges calculations and discussions with David Parfitt. The communications with Federico Baiutti and Albert Tarancon are also acknowledged. Open access fee was paid from the Imperial College London Open Access Fund.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Steele, B.C.H.; Heinzel, A. Materials for fuel-cell technologies. Nature 2001, 414, 345–352. [Google Scholar] [CrossRef]
- Tarascon, J.M. Key challenges in future Li-battery research. Philos. Trans. R. Soc. Lond. A 2010, 368, 3227–3241. [Google Scholar] [CrossRef]
- Brett, D.J.L.; Atkinson, A.; Brandon, N.P.; Skinner, S.J. Intermediate temperature solid oxide fuel cells. Chem. Soc. Rev. 2008, 37, 1568–1578. [Google Scholar] [CrossRef]
- Tarancón, A.; Burriel, M.; Santiso, J.; Skinner, S.J.; Kilner, J.A. Advances in layered oxide cathodes for intermediate temperature solid oxide fuel cells. J. Mater. Chem. 2010, 20, 3799–3813. [Google Scholar] [CrossRef]
- Kushima, A.; Yildiz, B. Oxygen ion diffusivity in strained yttria stabilized zirconia: Where is the fastest strain? J. Mater. Chem. 2010, 20, 4809–4819. [Google Scholar] [CrossRef]
- Souza, R.A.D.; Ramadan, A.; Hörner, S. Modifying the barriers for oxygen-vacancy migration in fluorite-structured CeO2 lectrolytes through strain: A computer simulation study. Energy Environ. Sci. 2012, 5, 5445–5453. [Google Scholar] [CrossRef]
- Rushton, M.J.D.; Chroneos, A. Impact of uniaxial strain and doping on oxygen diffusion in CeO2. Sci. Rep. 2014, 4, 6068. [Google Scholar] [CrossRef]
- Chiabrera, F.; Garbayo, I.; López-Conesa, L.; Martín, G.; Ruiz-Caridad, A.; Walls, M.; Ruiz-González, L.; Kordatos, A.; Núñez, M.; Morata, A.; et al. Engineering transport in manganites by tuning local nonstoichiometry in grain boundaries. Adv. Mater. 2019, 31, 1805360. [Google Scholar] [CrossRef]
- Baiutti, F.; Chiabrera, F.; Acosta, M.; Diercks, D.; Parfitt, D.; Santiso, J.; Wang, X.; Cavallaro, A.; Morata, A.; Wang, H.; et al. A high-entropy manganite in an ordered nanocomposite for long-term application in solid oxide cells. Nat. Commun. 2021, 12, 2660. [Google Scholar] [CrossRef]
- Rost, C.M.; Sachet, E.; Borman, T.; Moballegh, A.; Dickey, E.C.; Hou, D.; Jones, J.L.; Curtarolo, S.; Maria, J.-P. Entropy-stabilized oxides. Nat. Commun. 2015, 6, 8485. [Google Scholar] [CrossRef]
- Yeh, J.-W.; Chen, S.-K.; Lin, S.-J.; Gan, J.-Y.; Chin, T.-S.; Shun, T.-T.; Tsau, C.-H.; Chang, S.-Y. Nanostructured high-entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes. Adv. Eng. Mater. 2004, 6, 299–303. [Google Scholar] [CrossRef]
- Gali, A.; George, E.P. Tensile properties of high- and medium-entropy alloys. Intermetallics 2013, 39, 74–78. [Google Scholar] [CrossRef]
- Gludovatz, B.; Hohenwarter, A.; Catoor, D.; Chang, E.H.; George, E.P.; Ritchie, R.O. A fracture-resistant high-entropy alloy for cryogenic applications. Science 2014, 345, 1153–1158. [Google Scholar] [CrossRef]
- Bonnet, E.; Grenier, J.C.; Bassat, J.M.; Jacob, A.; Delatouche, B.; Bourdais, S. On the ionic conductivity of some zirconia-derived high-entropy oxides. J. Eur. Ceram. Soc. 2021, 41, 4505–4515. [Google Scholar] [CrossRef]
- Gild, J.; Samiee, M.; Braun, J.L.; Harrington, T.; Vega, H.; Hopkins, P.E.; Vecchio, K.; Luo, J. High-entropy fluorite oxides. J. Eur. Ceram. Soc. 2018, 38, 3578–3584. [Google Scholar] [CrossRef]
- Sarkar, A.; Wang, Q.; Schiele, A.; Chellali, M.R.; Bhattacharya, S.S.; Wang, D.; Brezesinski, T.; Hahn, H.; Velasco, L.; Breitung, B. High-entropy oxides: Fundamental aspects and electrochemical properties. Adv. Mater. 2019, 31, 1806236. [Google Scholar] [CrossRef]
- Akrami, S.; Edalati, P.; Fuji, M.; Edalati, K. High-entropy ceramics: Review of principles, production and applications. Mater. Sci. Eng. R Rep. 2021, 146, 100644. [Google Scholar] [CrossRef]
- Chen, K.P.; Pei, X.T.; Tang, L.; Cheng, H.R.; Li, Z.M.; Li, C.W.; Zhang, X.W.; An, L.A. A five-component entropy-stabilized fluorite oxide. J. Eur. Ceram. Soc. 2018, 38, 4161–4164. [Google Scholar] [CrossRef]
- Chellali, M.R.; Sarkar, A.; Nandam, S.H.; Bhattacharya, S.S.; Breitung, B.; Hahn, H.; Velasco, L. On the homogeneity of high entropy oxides: An investigation at the atomic scale. Scipta Mater. 2019, 166, 58–63. [Google Scholar] [CrossRef]
- Wright, A.J.; Wang, Q.Y.; Huang, C.Y.; Nieto, A.; Chen, R.K.; Luo, J. From high-entropy ceramics to compositionally-complex ceramics: A case study of fluorite oxides. J. Eur. Ceram. Soc. 2020, 40, 2120–2129. [Google Scholar] [CrossRef]
- Dabrowa, J.; Szymczak, M.; Zajusz, M.; Mikula, A.; Mozdzierz, M.; Berent, K.; Wytrwal-Sarna, M.; Bernasik, A.; Stygar, M.; Swierczek, K. Stabilizing fluorite structure in ceria-based high-entropy oxides: Influence of Mo addition on crystal structure and transport properties. J. Eur. Ceram. Soc. 2020, 40, 5870–5881. [Google Scholar] [CrossRef]
- Spiridigliozzi, L.; Ferone, C.; Cioffi, R.; Dell’Agli, G. A simple and effective predictor to design novel fluorite-structured High Entropy Oxides (HEOs). Acta. Mater. 2021, 202, 181–189. [Google Scholar] [CrossRef]
- Chen, K.P.; Ma, J.X.; Tan, C.A.X.; Li, C.W.; An, L.A. An anion-deficient high-entropy fluorite oxide with very low density. Ceram. Inter. 2021, 47, 21207–21211. [Google Scholar] [CrossRef]
- Su, L.; Chen, X.; Xu, L.; Eldred, T.; Smith, J.; DellaRova, C.; Wang, H.J.; Gao, W.P. Visualizing the formation of high-entropy fluorite oxides from an amorphous precursor at atomic resolution. ACS Nano 2022, 16, 21397–21496. [Google Scholar] [CrossRef]
- Nundy, S.; Tatar, D.; Kojcinovic, J.; Ullah, H.; Chosh, A.; Mallick, T.K.; Meinusch, R.; Smarsly, B.M.; Tahir, A.A.; Djerdj, I. Bandgap engineering in novel fluorite-type rare earth high-entropy oxides (RE-HEOs) with computational and experimental validation for photocatalytic water splitting applications. Adv. Sustain. Syst. 2022, 6, 2200067. [Google Scholar] [CrossRef]
- Kotsonis, G.N.; Almishal, S.S.I.; Vieira, F.M.D.; Crespi, V.H.; Dabo, I.; Rost, C.M.; Maria, J.P. High-entropy oxides: Harnessing crystalline disorder for emergent functionality. J. Am. Ceram. Soc. 2023, 106, 5587–5611. [Google Scholar] [CrossRef]
- Ma, B.; Wen, Z.Q.; Qin, J.D.; Wu, Z.Y.; Liu, J.X.; Lv, Y.M.; Yu, J.J.; Zhao, Y.H. Synthesis and microstructure of (Ce0.2Zr0.2La0.2Sm0.2Nd0.2)O2-δ high-entropy oxides characterized by fluorite structure. Ceram. Inter. 2024, 50, 1981–1989. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum Scales. Comp. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Buckingham, R.A.; Lennard-Jones, J.E. The classical equation of state of gaseous helium, neon and argon. Proc. R. Soc. London A 1938, 168, 264–283. [Google Scholar]
- Grimes, R.W.; Busker, G.; McCoy, M.A.; Chroneos, A.; Kilner, J.A.; Chen, S.P. The Effect of Ion Size on Solution Mechanism and Defect Cluster Geometry. Ber. Bunsenges. Phys. Chem. 1997, 101, 1204. [Google Scholar] [CrossRef]
- Rupasov, D.; Chroneos, A.; Parfitt, D.; Kilner, J.A.; Grimes, R.W.; Istomin, S.Y.; Antipov, E.V. Oxygen diffusion in Sr0.75Y0.25CoO2.625: A molecular dynamics study. Phys. Rev. B 2009, 79, 172102. [Google Scholar] [CrossRef]
- Ebmeyer, W.; Dholabhai, P.P. High-throughput prediction of oxygen vacancy defect migration near misfit dislocations in SrTiO3/BaZrO3 heterostructures. Mater. Adv. 2024, 5, 315–328. [Google Scholar]
- Gale, J.D. GULP: Capabilities and prospects. Z. Für Krist.-Cryst. Mater. 2005, 220, 552–554. [Google Scholar] [CrossRef]
- Gale, J.D. GULP: A Computer Program for the Symmetry-Adapted Simulation of Solids. J. Chem. Soc. Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef]
- Kushima, A.; Parfitt, D.; Chroneos, A.; Yildiz, B.; Kilner, J.A.; Grimes, R.W. Interstitialcy diffusion of oxygen in tetragonal La2CoO4+δ. Phys. Chem. Chem. Phys. 2011, 13, 2242–2249. [Google Scholar] [CrossRef]
- He, X.; Zhu, X.; Epstein, A.; Mo, Y. Statistical variances of diffusional properties from ab initio molecular dynamics simulations. npj Comput. Mater. 2018, 4, 18. [Google Scholar] [CrossRef]
- Varotsos, P.; Alexopoulos, K. Thermodynamics of Point Defects and their Relation with the Bulk Properties; North-Holland: Amsterdam, The Netherlands, 1986. [Google Scholar]
- Cooper, M.W.D.; Grimes, R.W.; Fitzpatrick, M.E.; Chroneos, A. Modeling oxygen self-diffusion in UO2 under pressure. Solid State Ionics 2015, 282, 26–30. [Google Scholar] [CrossRef]
- Chroneos, A. Connecting point defect parameters with bulk properties to describe diffusion in solids. Appl. Phys. Rev. 2016, 3, 041304. [Google Scholar] [CrossRef]
Figure 1.
Oxygen ion diffusivities and calculated activation energies for two ESOs HEO_A, Zr(1−x)/3Hf(1−x)/3Ce(1−x)/3Gdx/2Yx/2O2−x, HEO_B Zr(1−x)/3Hf(1−x)/3Ce(1−x)/3Yx/2Lax/2O2−x at x = 0.14 (8%RE2O3) compared with predicted oxygen ion diffusivity for Z8Y and C8Y.
Figure 1.
Oxygen ion diffusivities and calculated activation energies for two ESOs HEO_A, Zr(1−x)/3Hf(1−x)/3Ce(1−x)/3Gdx/2Yx/2O2−x, HEO_B Zr(1−x)/3Hf(1−x)/3Ce(1−x)/3Yx/2Lax/2O2−x at x = 0.14 (8%RE2O3) compared with predicted oxygen ion diffusivity for Z8Y and C8Y.

Figure 2.
The plots show (top) the overall diffusivity of the three HEOs and Z8Y as a function of the doping concentration together with (middle) the activation energy and (bottom) the pre-exponent. In each plot, the colored points show the individual data points and the solid lines show the data fitted to the activation energy and pre-exponent.
Figure 2.
The plots show (top) the overall diffusivity of the three HEOs and Z8Y as a function of the doping concentration together with (middle) the activation energy and (bottom) the pre-exponent. In each plot, the colored points show the individual data points and the solid lines show the data fitted to the activation energy and pre-exponent.
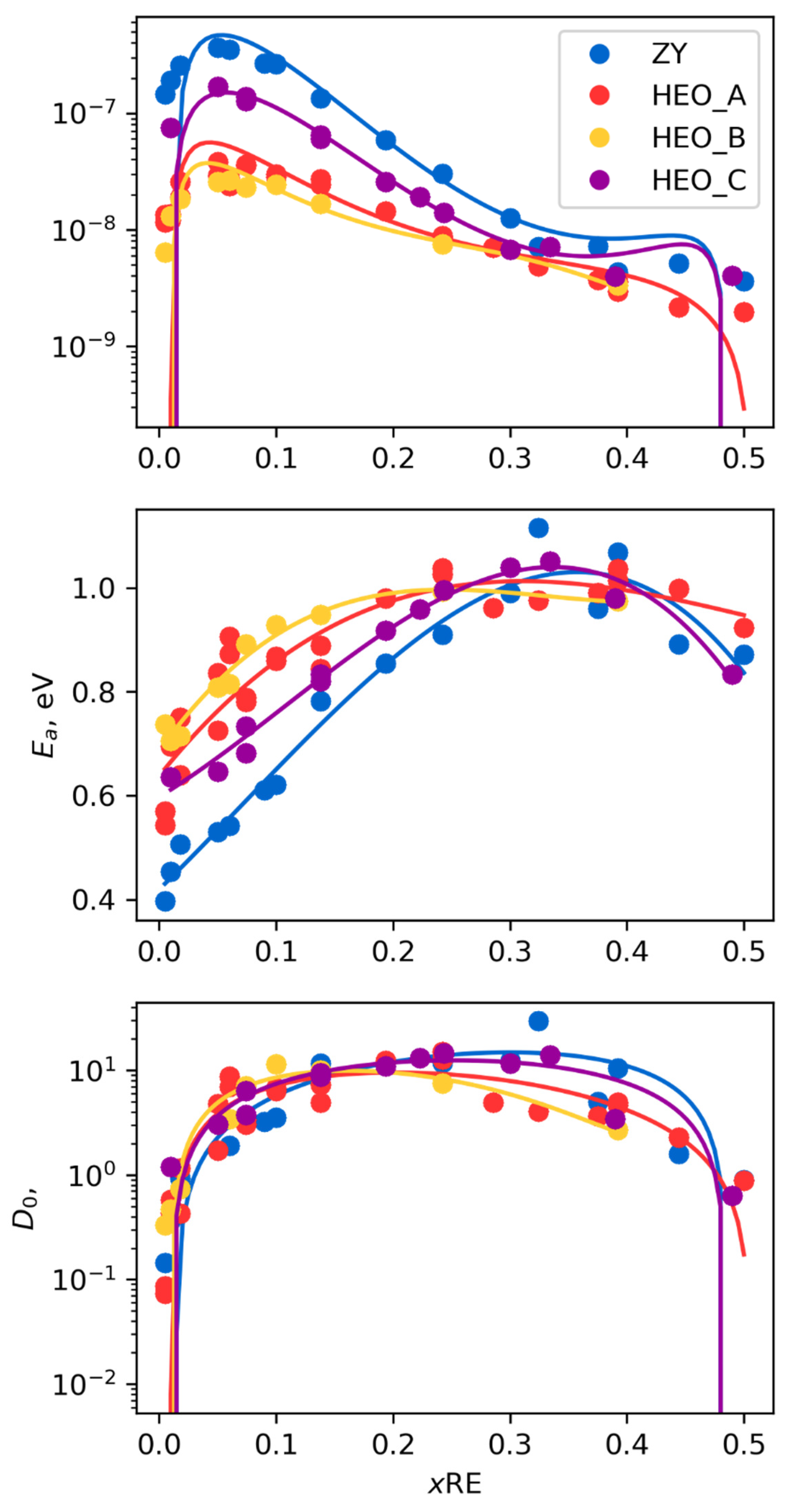
Figure 3.
Plot showing (a) the fluorite structure with metal ions colored blue (Y) or black (Ce, Hf, and Zr) and oxygen ions as red. An oxygen vacancy exists at (1/4, 1/4, 1/4), which, in this example, is adjacent to a Y3+ ion. The calculated transition path (b) in moving this vacancy to the next nearest neighbor position, i.e., to (3/4, 1/4, 1/4). The overall transition barrier Ets is split into a reaction energy Er and a barrier energy Eb.
Figure 3.
Plot showing (a) the fluorite structure with metal ions colored blue (Y) or black (Ce, Hf, and Zr) and oxygen ions as red. An oxygen vacancy exists at (1/4, 1/4, 1/4), which, in this example, is adjacent to a Y3+ ion. The calculated transition path (b) in moving this vacancy to the next nearest neighbor position, i.e., to (3/4, 1/4, 1/4). The overall transition barrier Ets is split into a reaction energy Er and a barrier energy Eb.
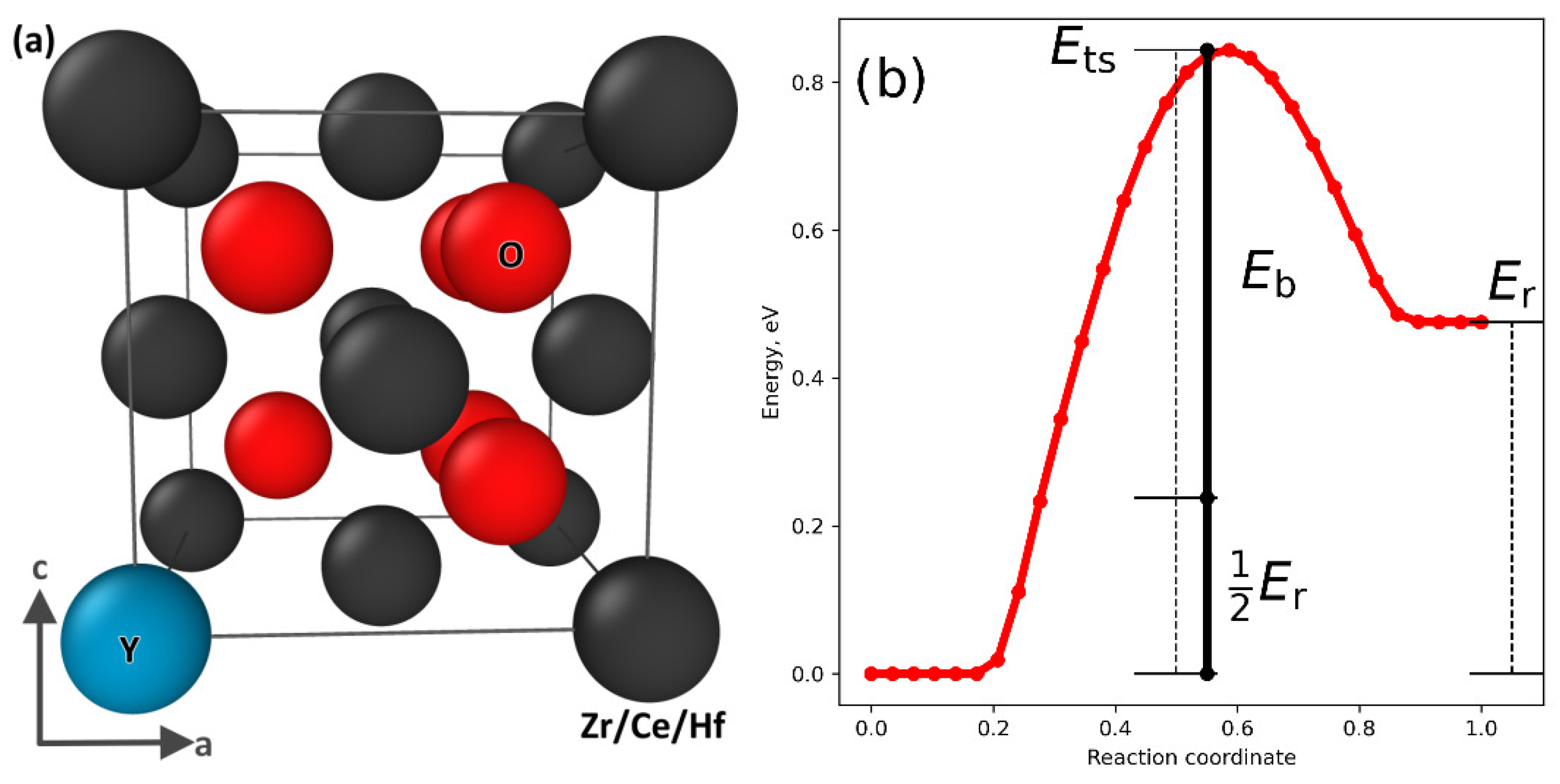
Figure 4.
Box and whisker diagram of the population of (a) reaction energies (Er) and (b) barrier energies for oxygen vacancy diffusion for 6.25% Y doping in ZrO2 with progressively greater amount of Ce and Hf additions.
Figure 4.
Box and whisker diagram of the population of (a) reaction energies (Er) and (b) barrier energies for oxygen vacancy diffusion for 6.25% Y doping in ZrO2 with progressively greater amount of Ce and Hf additions.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chroneos, A. Oxygen Self-Diffusion in Fluorite High Entropy Oxides. Appl. Sci. 2024, 14, 5309. https://doi.org/10.3390/app14125309
AMA Style
Chroneos A. Oxygen Self-Diffusion in Fluorite High Entropy Oxides. Applied Sciences. 2024; 14(12):5309. https://doi.org/10.3390/app14125309
Chicago/Turabian StyleChroneos, Alexander. 2024. "Oxygen Self-Diffusion in Fluorite High Entropy Oxides" Applied Sciences 14, no. 12: 5309. https://doi.org/10.3390/app14125309
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.